






![]()
================================
分享几篇关于熔化温度相关的文献:
这些文献主要聚焦单相方法、过热–过冷方法、Z 方法、修正 Z 方法、孔洞方法、修正孔洞方法、两相方法、夹层方法以及修正两相方法的准确性和高效性展开了系统比较。将上述熔化模型归为完美晶体、缺陷晶体以及固–液界面三类。
-
这些文献讨论了原子数目、升温速率、晶体缺陷、表面能对熔化温度的影响,以及熔化过程中局域原子结构的转变和原子的动力学行为;同时也阐述了预熔化、表面熔化和体熔化之间的异同。
-
这些文献分析了熔化和结晶过程中由升温速率导致的过热极限温度和冷却速率引起的非晶转变温度。
-
这些文献本研究详细探讨了均匀形核、非均匀形核、固相和液相的不同占比、固–液界面能以及固–液界面的数量对熔化行为的驱动程度。
================================
第1篇
================================
关键词:
1. Spherical defect/spherical void
2. Embedded atom method
3. Molecular dynamics
4. Bond-orientational order parameters
5. Voronoi tessellation
6. Velocity auto-correlation functions
7. Melting
================================
主要内容
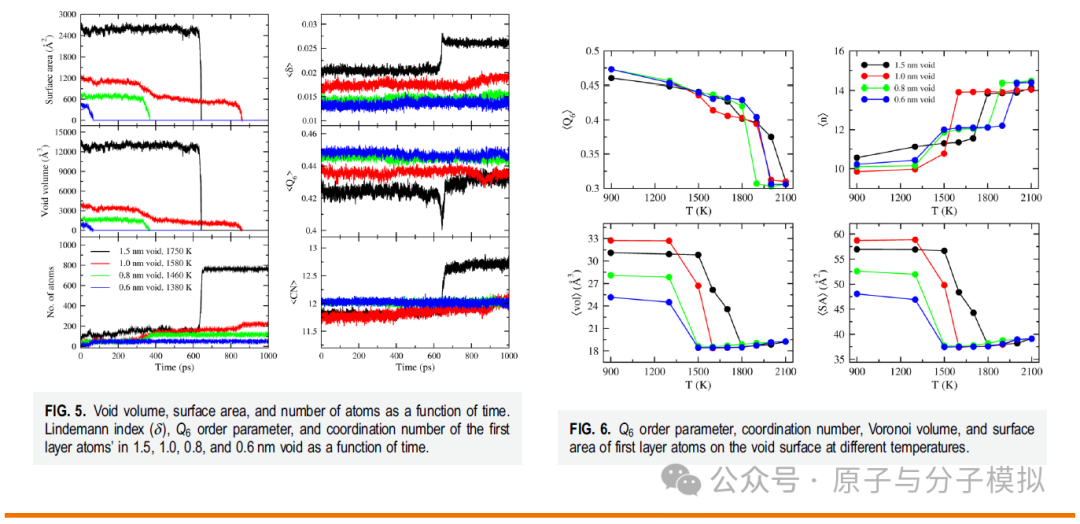
“钛 (Ti) 是多种工业应用中最重要的金属之一,球形缺陷的存在会降低其强度和稳定性。我们模拟了具有半径为 0.6、0.8、1.0 和 1.5 nm 的球形空隙的钛晶体以及没有球形空隙的晶体的熔化。Ti 采用嵌入原子法建模,所有晶体在 1 个大气压下从 300 K 加热到 2200 K 直至完全熔化。所有分子动力学轨迹均使用径向分布函数、键取向有序参数、Voronoi 和速度自相关函数进行分析。结果表明,0.6、0.8、1.0 和 1.5 nm 的空隙在晶体熔化之前被填充,并且在几皮秒内立即填充;此后,原子重新排列/排序为晶体状排列,其中对于具有0.6和0.8nm空隙的晶体,总体结晶度保持hcp,而对于具有1.0和1.5nm空隙的晶体,整体结晶度改变为bcc。对于所有有或没有空隙的晶体,熔化都是在长程和短程有序都丢失的情况下发生的,而不是像经典成核理论所提出的那样从液体状的原子核中发生。”——取自文章摘要。
================================


================================
Figure 1
Figure 2
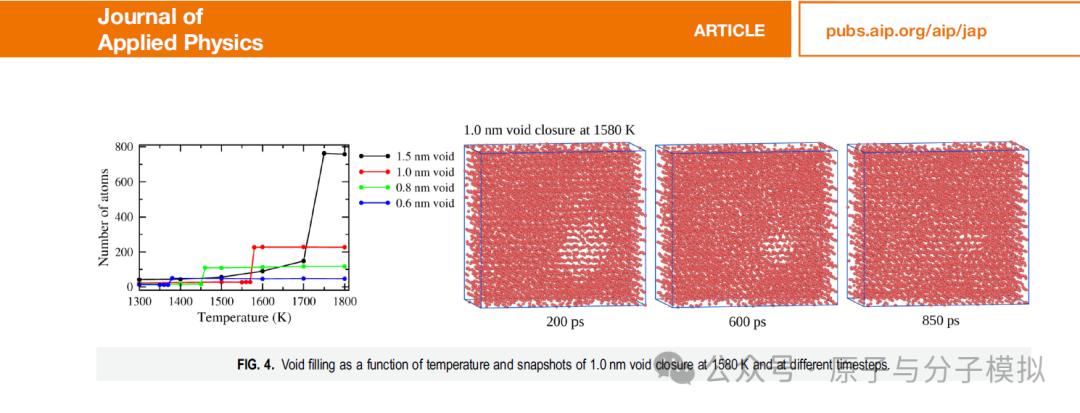
Figure 3
文章题目:
Understanding melting of Ti crystals with spherical voids from molecular dynamics simulations
文章链接:
https://doi.org/10.1063/5.0186850
================================
第2篇
================================
文章题目:
Molecular dynamics simulation of homogeneous nucleation of melting in superheated sodium crystal
文章链接:
https://doi.org/10.1016/j.jcrysgro.2023.127460

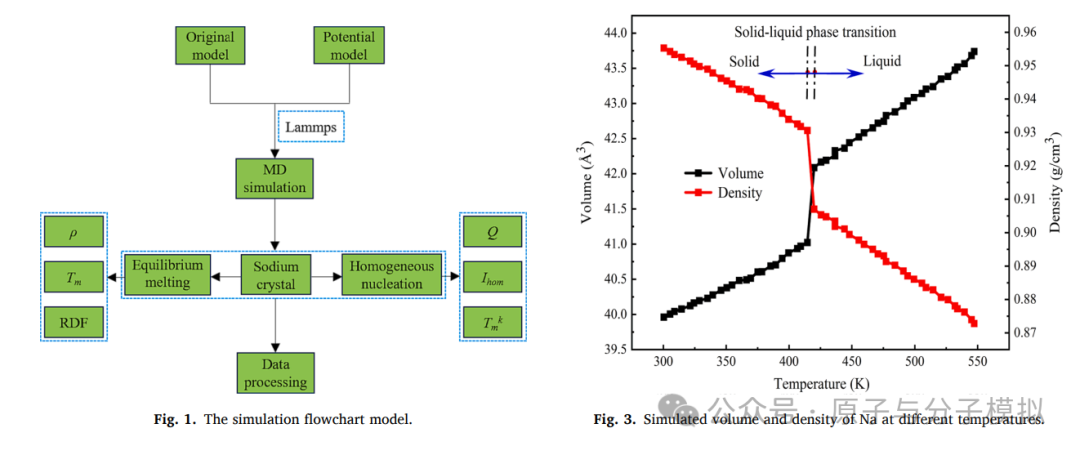
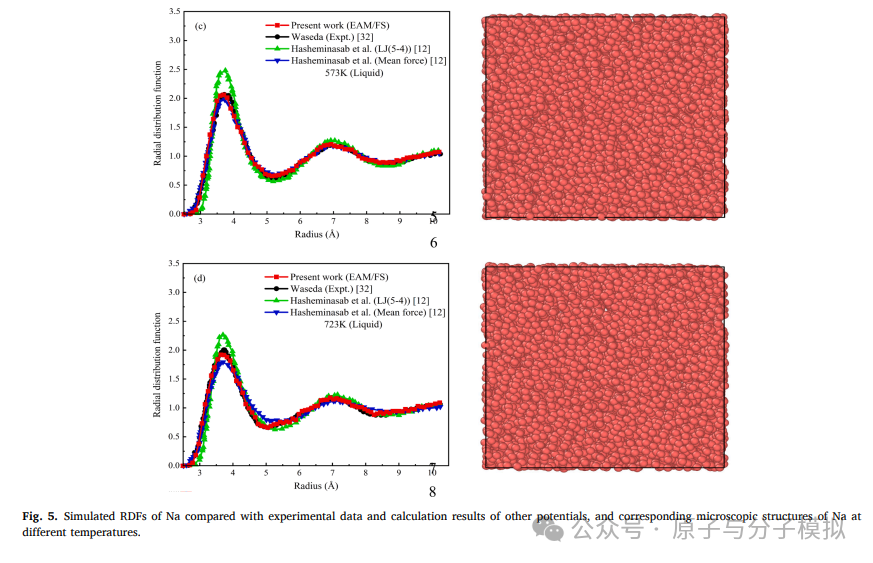
第3篇
================================
文章题目:
A comprehensive investigation on the accuracy and efficiency of methods for melting temperature calculation using molecular dynamics simulations
文章链接:
https://doi.org/10.1016/j.molliq.2023.123924

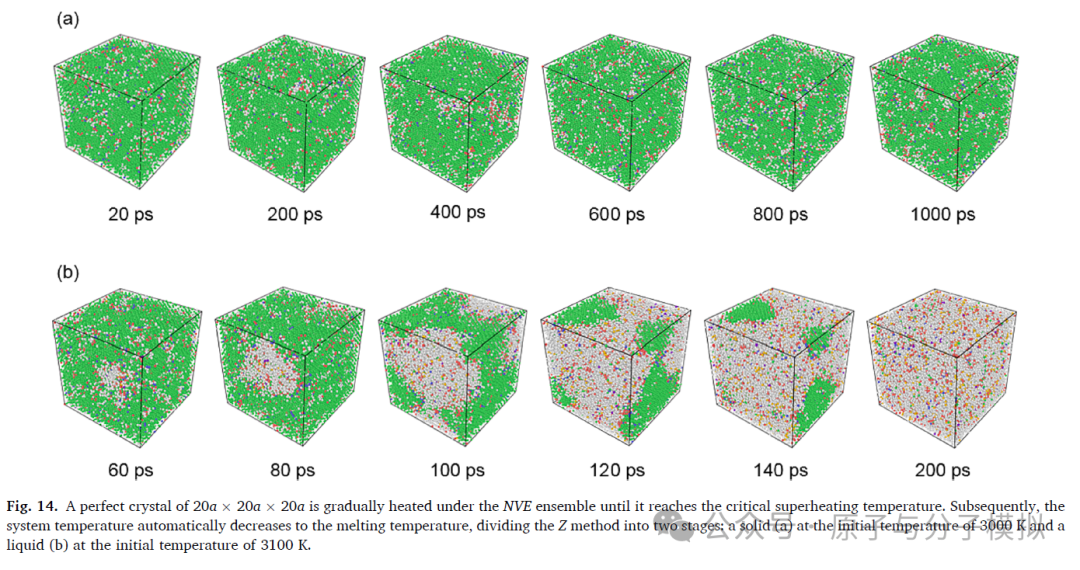
第4篇
================================
文章题目:
Simulation and analysis of the local atomic structure for melting behavior in metals
文章链接:
https://doi.org/10.1016/j.physb.2024.415747

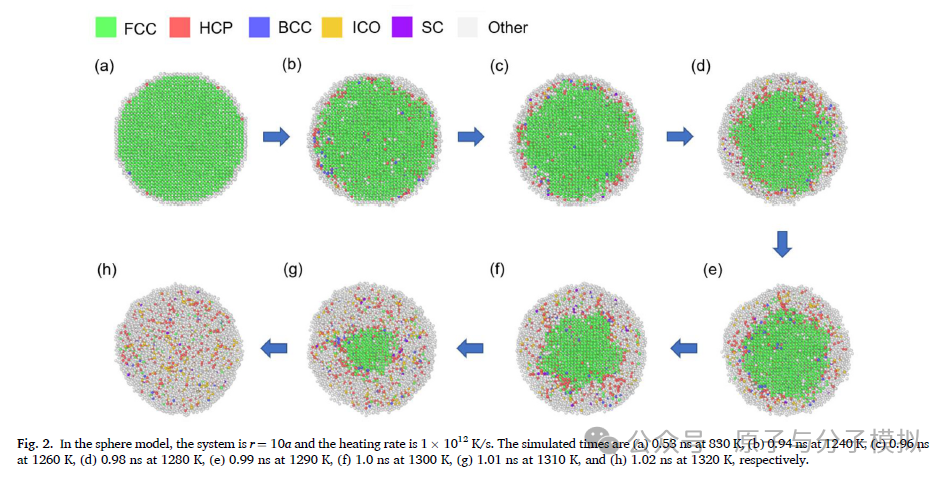
第5篇
================================
文章题目:
Partial disordering and homogeneous melting in multicomponent systems
文章链接:
https://doi.org/10.1016/j.actamat.2022.118281

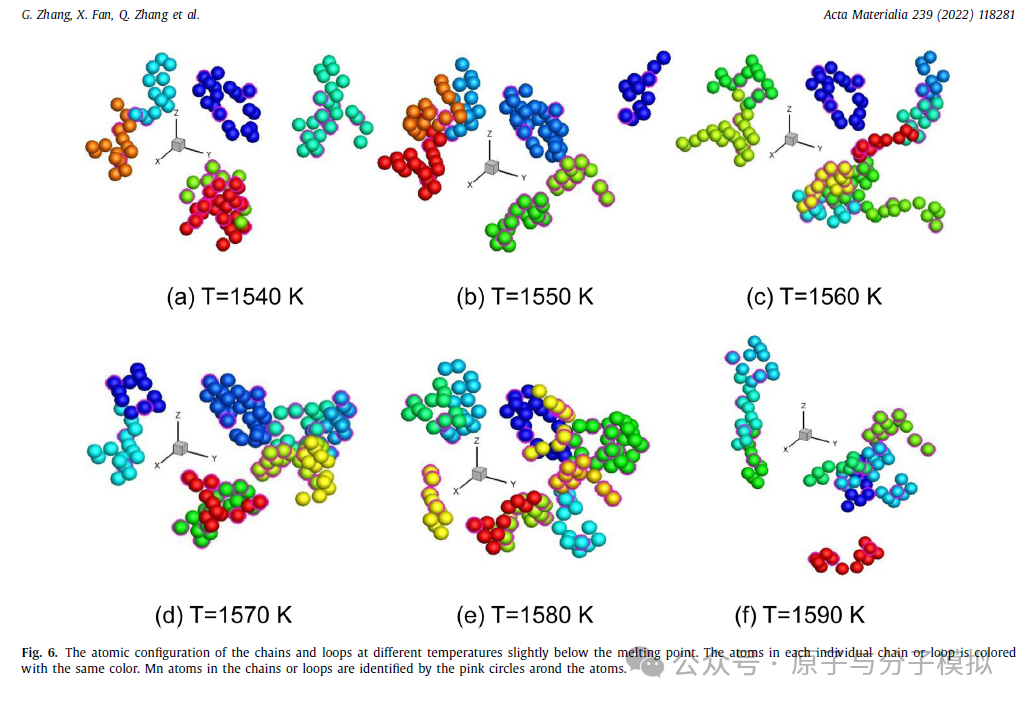
第6篇
================================
文章题目:
Localization and delocationzation of surface disordering in surface mediated melting
文章链接:
DOI: 10.1103/PhysRevB.104.134204

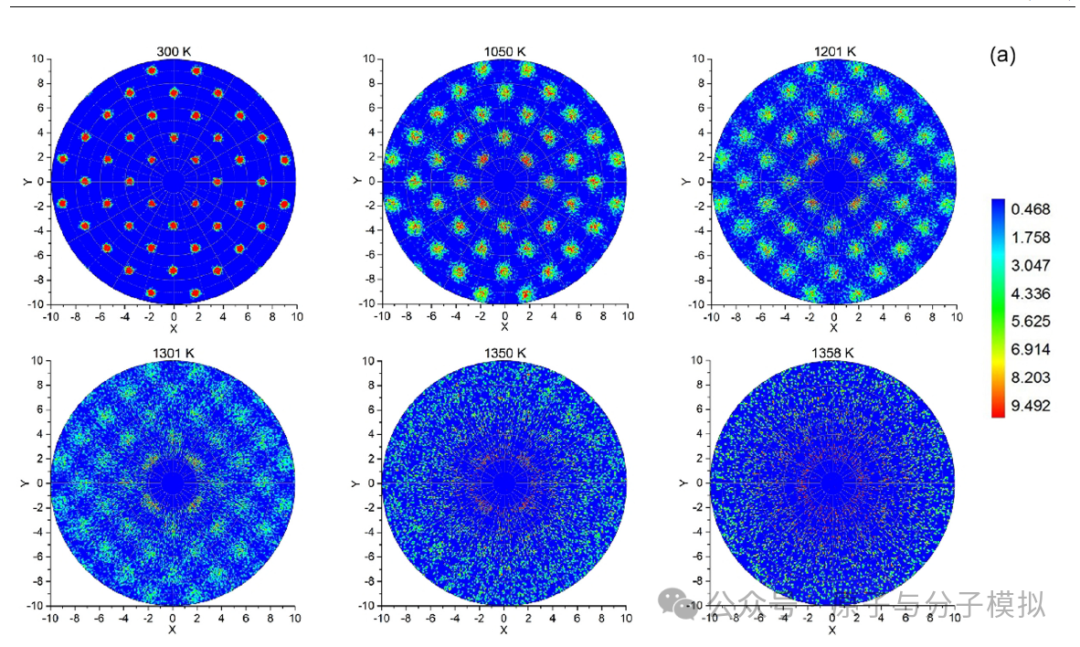
第7篇
================================
文章题目:
Molecular dynamics simulation of the crystal structure evolution of titanium under different Tdamp values and heating/cooling rates
文章链接:
https://doi.org/10.1016/j.cplett.2020.138187

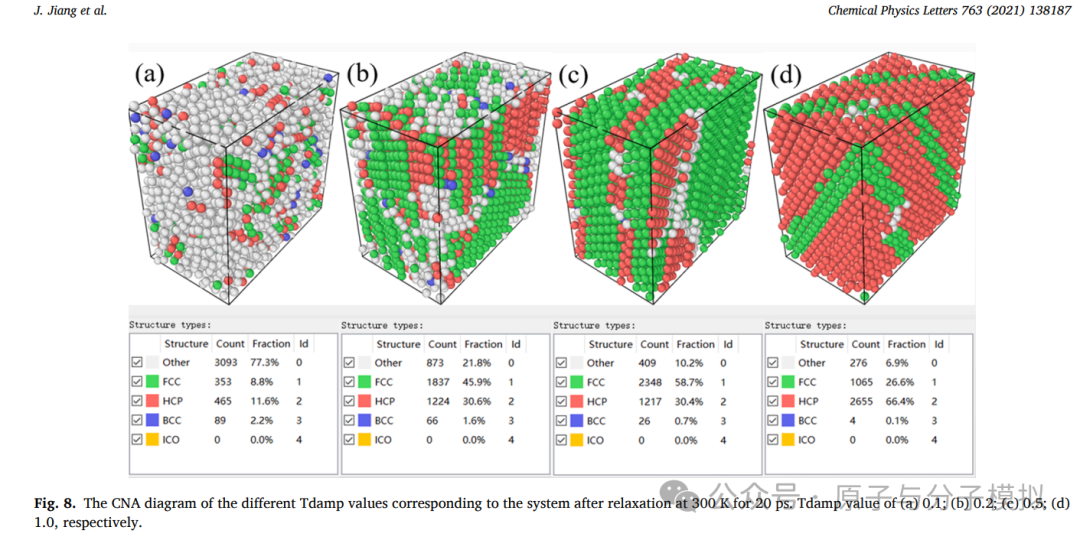
第8篇
================================
文章题目:
Investigation on the efficiency and accuracy of methods for calculating melting temperature by molecular dynamics simulation
文章链接:
https://doi.org/10.1016/j.commatsci.2019.109156

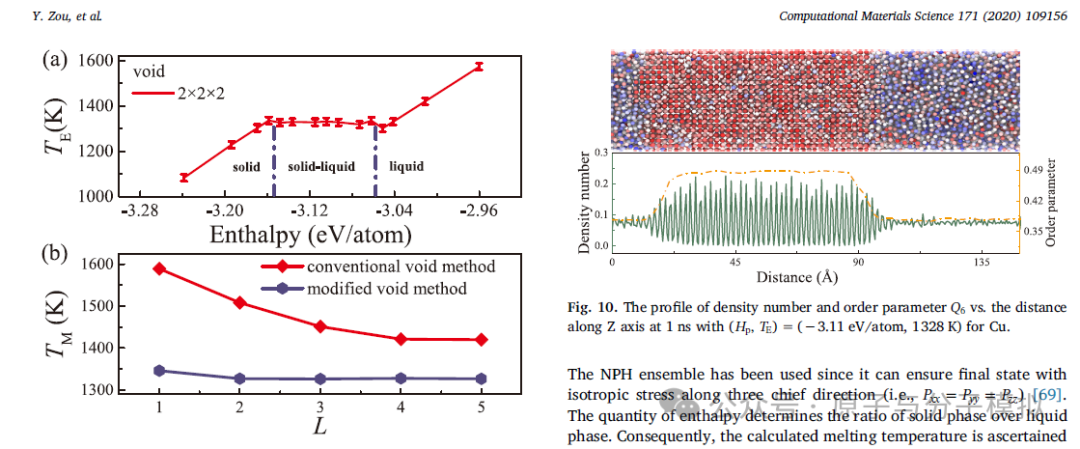
第9篇
================================
文章题目:
Rethinking Lindemann criterion: A molecular dynamics simulation of surface mediated melting
文章链接:
https://doi.org/10.1016/j.actamat.2020.05.013

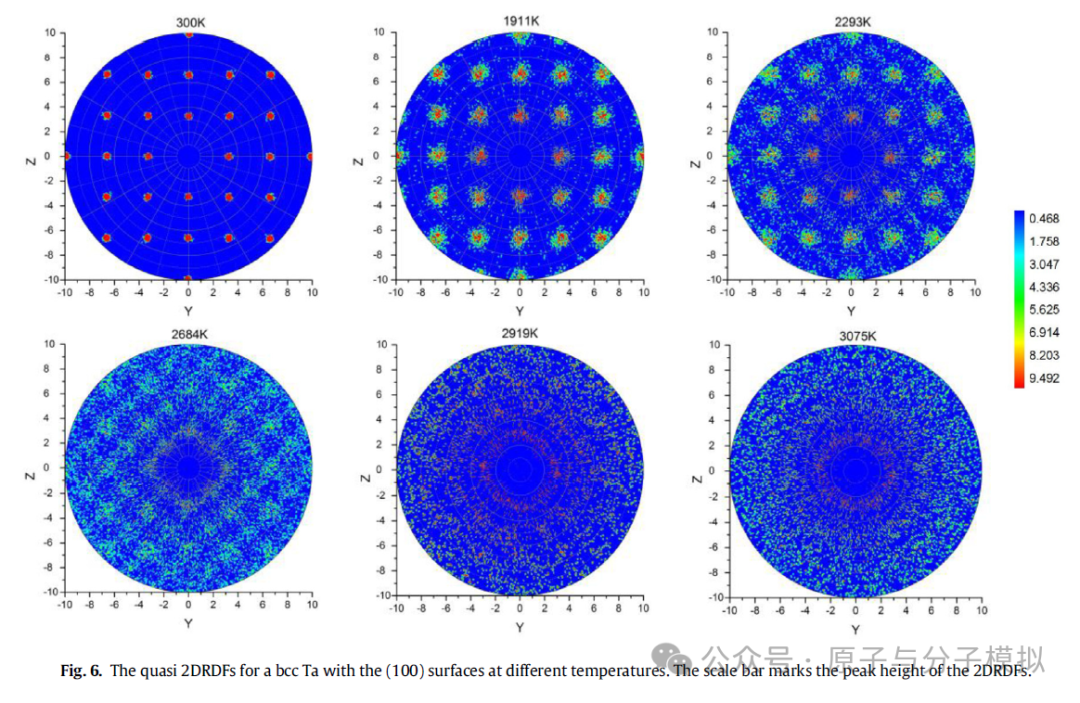
第10篇
================================
文章题目:
Study of structural stability of copper crystal with voids from molecular dynamics simulations
文章链接:
https://doi.org/10.1016/j.cplett.2019.06.046


第11篇
================================
文章题目:
Size effect on melting temperatures of alumina nanocrystals: Molecular dynamics simulations and thermodynamic modeling
文章链接:
https://doi.org/10.1016/j.commatsci.2017.12.064

================================
![]()
以上是我们分享的一些经验或者文章的搬运,或有不足,欢迎大家指出!
如有侵权,请联系我立马删除!
详细内容(文章题目、文章链接、附件下载)可在微 信 公 众 号:原子与分子模拟获取,欢迎大家关注。


















 4365
4365

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









