




蛋白质作为生命活动的主要执行者,其功能往往通过多链复合物的形式实现。从抗体-抗原识别到酶-底物结合,多链蛋白质间的精确相互作用是理解生命机制的核心。然而,当前 AI 驱动的蛋白质建模领域呈现出显著的「单链偏向性」,虽然 AlphaFold 、 ESM 系列等模型已在单链蛋白质的折叠与设计中取得突破性进展,但多链复合物的建模仍处于起步阶段。
现有方法处理多链蛋白质普遍采用「伪序列连接」策略,将多链强制视为单链处理。这种方法严重限制了链间相互作用的自然表达——真实生物复合物中,链间空间位置与结合界面的原子级相互作用(如氢键、疏水作用)无法通过线性连接准确建模。此外,全原子结构的生成面临双重挑战:氨基酸侧链的复杂构象与序列-结构的强依赖性,使得多链复合物的从头设计成为领域难题。
为了填补这一研究空白,湖南大学联合中国科学院大学、字节跳动 Seed 团队提出 APM(All-Atom Protein Generative Model),一款专为多链蛋白质复合物设计的全原子蛋白质生成模型。 APM 不仅能直接生成具有全原子结构的多链复合物,还支持折叠、逆折叠等基础任务,并在抗体、肽等功能蛋白设计中展现出卓越性能。
研究成果以「An All-Atom Generative Model for Designing Protein Complexes」为题,入选 ICML 2025 。
研究亮点:
* 多链原生建模:摒弃伪序列连接,直接学习多链独立空间分布与结合界面的原子级相互作用;
* 全原子表示优化:平衡计算效率与结构细节,通过氨基酸类型、 backbone 框架与侧链扭转角的联合表示,实现原子级结构生成;
* 序列-结构依赖强化:通过解耦噪声过程与双向任务训练(折叠/逆折叠),维持序列与结构的深层关联。

论文地址:
An All-Atom Generative Model for Designing Protein Complexes | OpenReview
关注公众号,后台回复「APM」获取完整 PDFAPM 蛋白质生成数据集:
APM 蛋白质生成数据集 | 数据集 | HyperAI超神经
更多 AI 前沿论文:
数据集:从单链到多链的丰富样本
APM 的训练基于精心构建的多源蛋白质数据集,整合了单链与多链蛋白质的结构与序列信息,为模型提供了丰富的学习素材。
单链数据集通过多源融合与质量过滤为链内建模提供丰富基础,共包含 187,494 个样本,覆盖了广泛的蛋白质类型与功能类别其中,其数据主要来自 3 个权威数据库:
* PDB 数据库:经过 MultiFlow 数据处理流程,筛选得到 18,684 个样本;
* Swiss-Prot 数据库:选取 pLDDT>85 的高质量结构,获得 140,769 个样本;
* AFDB 数据库:采用更严格的筛选标准,保留 pLDDT>95 的样本,共计 28,041 个样本。
多链蛋白质数据集共包含 11,620 个样本,涵盖 2-6 条链的蛋白质复合物,为多链建模提供了关键数据支撑。多链蛋白质数据源自 PDB 生物组装数据(Biological Assemblies),为避免下游任务的信息泄露,研究团队排除了 3 类样本:在 SAbDab 抗体数据库中存在的样本;包含长度小于 30 的链(视为肽段)的样本;长度超过 2,048 或缺乏聚类 ID 的样本。
为提升模型的泛化能力,研究人员在训练过程中对多链样本进行了随机裁剪处理:对于残基数超过 384 的样本,以链间结合界面的残基对为中心,保留最近的 384 个氨基酸。这种裁剪策略确保模型能够聚焦于关键结合区域,同时避免内存溢出问题。此外,研究人员还将单链与多链数据按比例混合,利用单链数据的丰富性提升链内建模能力。每个采样位置都附加了丰富的元数据,包括地理位置(链间相互作用位点)、结构属性(如二级结构类型)、序列特征(氨基酸类型与保守性)。这些信息为模型学习序列-结构-功能的映射关系提供了多维度线索。
APM 蛋白质生成数据集:
APM 蛋白质生成数据集 | 数据集 | HyperAI超神经
模型架构:三模块协同的全原子生成框架
APM 的核心架构由序列与 backbone 生成模块(Seq&BB Module)、侧链生成模块(Sidechain Module)和全原子优化模块(Refine Module)3 个功能明确的模块协同组成,通过创新的设计实现了从序列到全原子结构的端到端生成,同时支持多链蛋白质的各种设计任务。
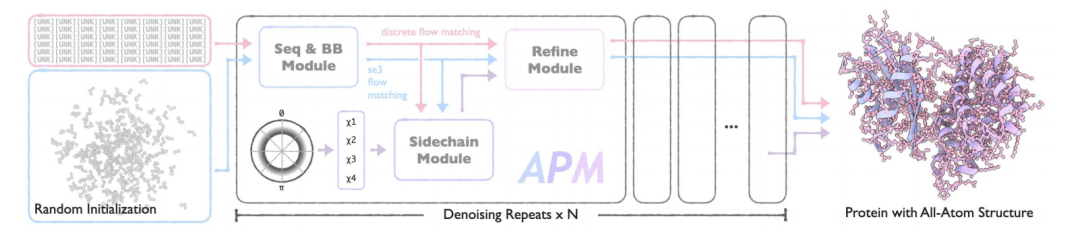
APM 核心架构图
Seq&BB Module
该模块是 APM 的基础,采用流匹配(Flow Matching)方法,实现序列与蛋白质 backbone 的联合生成,能够处理残基级别的序列-结构协同建模任务。通过解耦序列和结构的噪声过程,减少对序列-结构依赖关系的破坏,同时以 50% 概率执行折叠/逆折叠任务,强化双向依赖学习。模块的核心创新在于:
* 解耦噪声过程:将序列与结构的噪声过程分离,避免传统方法中模态间依赖关系的破坏。噪声序列与噪声 backbone 的时间步独立采样,确保模型能够学习双向的序列-结构依赖关系。
* SE(3) 流匹配:针对蛋白质 backbone 的空间变换特性,引入三维特殊欧几里得群(SE(3))流匹配,分别处理平移与旋转部分
* 多任务学习:同时支持无条件生成、条件生成、折叠和逆折叠任务,通过混合任务训练提升模型的泛化能力。损失函数包含流匹配损失和一致性损失,确保生成轨迹的平滑性。
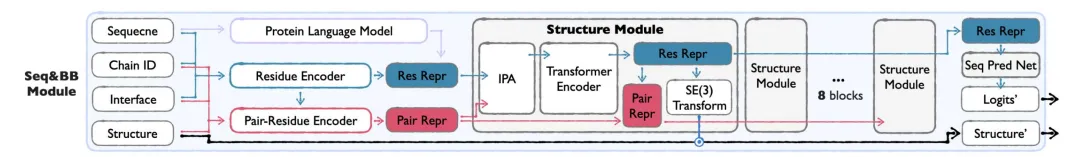
Seq&BB Module 架构图
Sidechain Module
为实现全原子结构生成,Sidechain Module 基于 Seq&BB 生成的序列与 backbone,预测氨基酸侧链的构象。
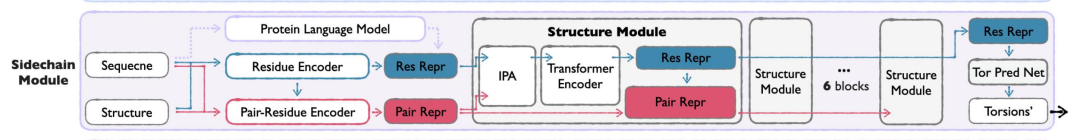
Sidechain Module 架构图
模块采用以下策略:
* torsion angle 表示:通过侧链扭转角(最多 4 个可旋转键)参数化侧链结构,平衡计算效率与原子级细节,避免直接建模全原子坐标带来的复杂度。
* 两阶段训练:第一阶段专注于侧链 packing 任务,学习真实侧链构象的分布;第二阶段切换为从预测结构重建真实侧链,确保模型在生成场景下的适用性。
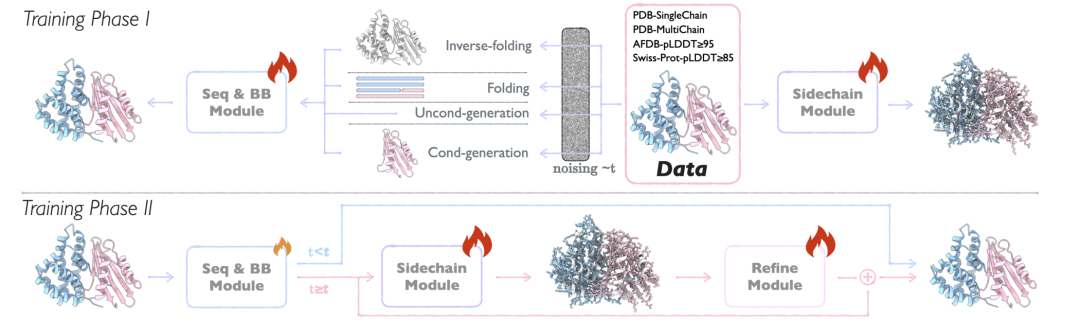
APM 训练过程 2 个阶段
* 轻量级设计:相比 Seq&BB Module,Sidechain Module 采用更少的结构块和更小的隐藏维度。
Refine Module
Refine Module 作为 APM 的最后一环,整合 Seq&BB 和 Sidechain Module 的输出,通过校正损失优化序列与 backbone,减少原子冲突并提升结构合理性。利用全原子信息优化序列和主链结构,解决结构冲突,使生成结果更接近天然蛋白质。该模块仅在生成后期(t≥0.8)激活,确保输入质量足以支撑优化。
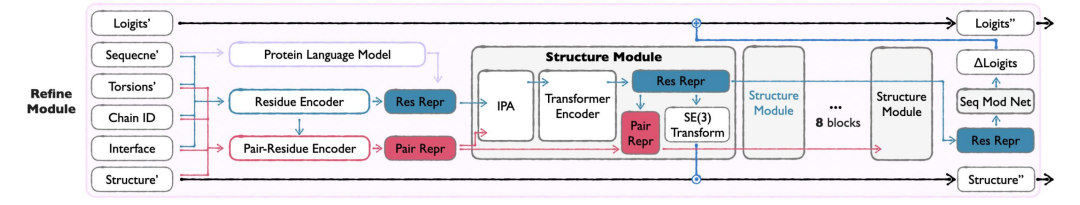
Refine Module
实验结论:多维度验证 APM 的突破性性能
APM 的实验验证覆盖单链基础任务、多链核心任务与下游功能设计,结果均表现优异。
单链蛋白质任务:可媲美专业模型的基础能力
折叠任务中,在 PDB 数据集上,APM 的 RMSD 为 4.83/2.64,TM-score 达 0.86/0.91,与 ESM3 、 MultiFlow 等模型性能相当;逆折叠任务中,氨基酸回收率(AAR)达 50.44%,超越 ProteinMPNN 的 46.58% 。
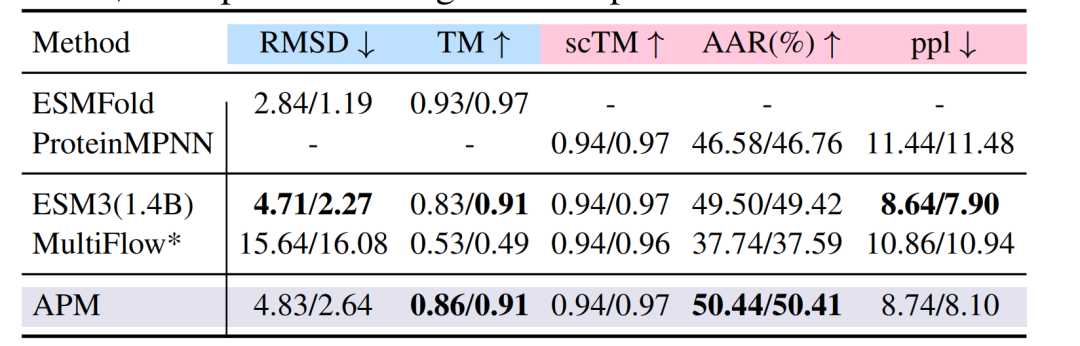
蛋白质折叠(蓝色高亮)和逆折叠任务(粉色高亮)的性能比较
此外,如下图所示,在无条件生成的长度为 100-300 的残基蛋白中,APM 的 scTM 达 0.96(Length 100),scRMSD 低至 1.80,显著优于 ESM3(1.4B)、 ProtPardelle 等全原子设计模型。
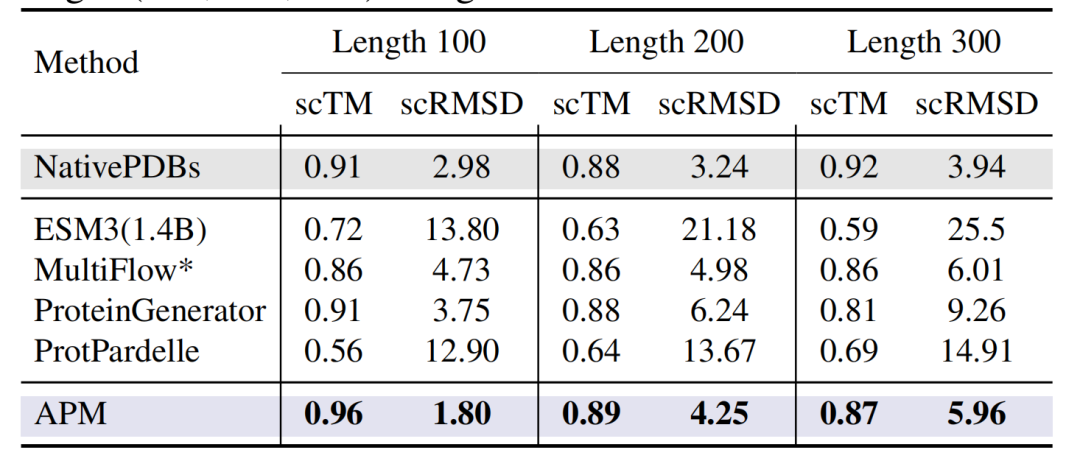
不同方法对不同蛋白质长度的性能比较
多链蛋白质任务:原生建模的核心优势
折叠与逆折叠的实验中,在 2-6 链复合物上,APM 的折叠性能为 12.6/13.67,虽低于 Boltz-1,但在无 MSA 条件下显著超越 Boltz-1;逆折叠的 scTM 达 0.85/0.95,接近带 MSA 的 Boltz-1,证明序列-结构关联的有效性。实验结果如下图所示。
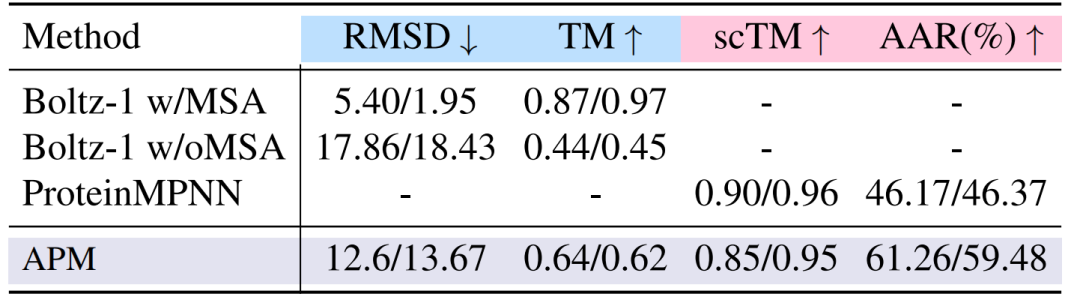
多链蛋白质折叠(蓝色突出显示)和逆折叠任务(粉色突出显示)的性能比较
其次,生成的多链复合物具有强结合亲和力,以 50-100 链长为例,全原子松弛后的结合能 ΔG_RAA 达 -112.65/-116.98,显著优于 Chroma(-83.96/-86.66)和仅用主链的 APM_BB(-114.94/-114.45),证明全原子信息对链间作用建模的必要性。
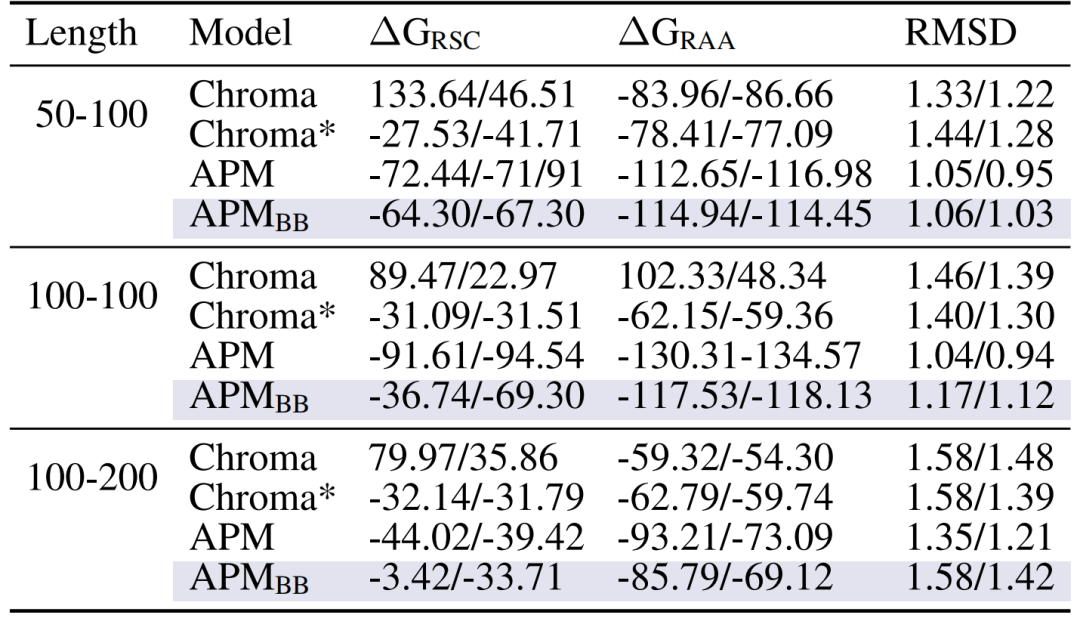
生成的复合物之间的链间结合亲和力
下游功能设计:抗体与肽段的应用突破
抗体 CDR-H3 设计:在 RAbD 基准测试中,APM 的 AAR 达 41.20%,RMSD 为 2.08,结合能 ΔG 为 91.64,全面超越 dyMEAN 、 DiffAb 等方法;零样本生成的抗体虽序列与天然差异大,但结合能更优(ΔG 81.12),证明其通用结合能力。
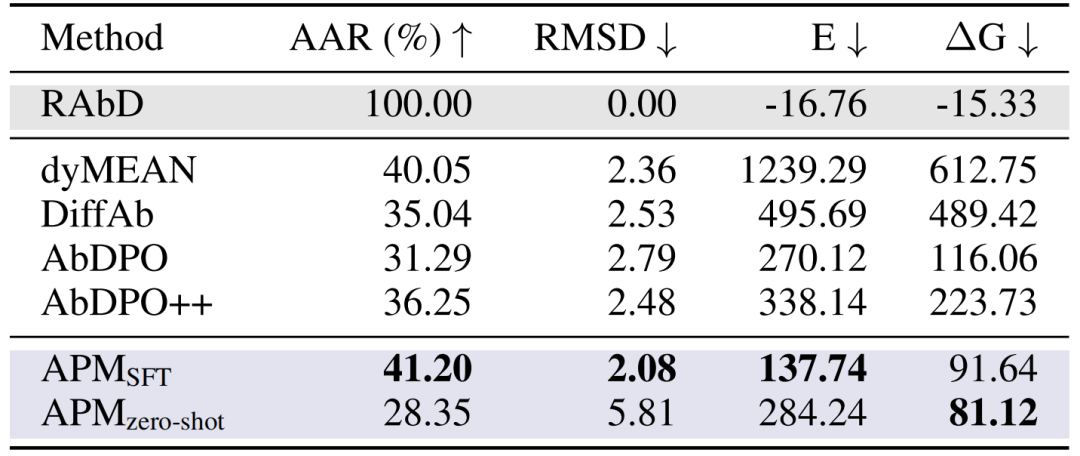
抗体设计方法在 RAbD 基准上的性能比较
肽段设计:在 PepBench 和 LNR 数据集上,研究人员从功能性、可折叠性和准确性三个关键方面对肽设计方法进行全面评估。如下图所示,APM(SFT)的结合能 ΔG 达 -19.90,69.34% 样本 ΔG<0,DockQ≥0.8 的比例达 11.29%,远超 PPFlow 、 PepGLAD 等方法,且折叠稳定性(pLDDT 60.36,ipTM 0.66)优异。
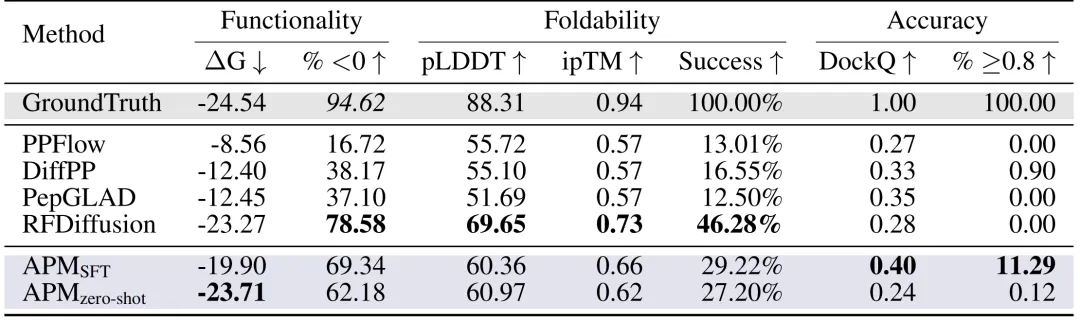
对肽设计方法的全面评估
产研协同驱动全原子蛋白质生成技术革新突破
在全原子蛋白质生成这一生物前沿领域,学术界与企业界对其探索从未停歇,一系列突破性成果持续引发关注。
学术界方面,DeepMind 团队推出的 AlphaFold3 在全原子蛋白质生成领域展现出强大能力,其通过融合多尺度结构信息与进化序列数据,实现了对复杂蛋白质折叠模式的精准建模,尤其在包含辅因子、金属离子的全原子复合体生成任务中,较传统方法在结构精度与能量合理性上均实现显著提升。斯坦福大学研究团队开发的 ESM-IF1 则另辟蹊径,基于巨量进化序列数据训练的隐式折叠模型,能直接生成具有天然构象特征的全原子蛋白质结构,在酶活性中心的精准构建上表现突出。
企业界同样在该领域积极布局,以技术创新推动产业应用。北京百奥几何生物科技有限公司发布了全球首个全场景原子级蛋白质大模型——GeoFlow V2,构建了端到端的扩散生成框架,可实现对蛋白质原子级别的精准调控,在抗体 CDR 区全原子设计中,能同时优化亲和力与稳定性,显著提升药物开发效率。美国生物科技公司英矽智能研发了一款蛋白质生成系统,聚焦药物靶点蛋白设计,其采用的多约束条件生成策略,可在保证全原子结构合理性的前提下,定向优化蛋白质与小分子药物的结合位点,为候选药物的高效筛选提供坚实基础。
这些学术界的理论突破与企业界的应用创新,共同推动全原子蛋白质生成技术从实验室走向产业实践,为精准药物研发、新型生物催化剂设计及合成生物学领域的突破提供了核心支撑,未来有望在疾病治疗与生物制造领域创造巨大价值。
参考链接:


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









