




 slingshot 是一个用于单细胞转录组数据轨迹推断的R包,通过构建最小生成树(MST)识别全局谱系结构,并通过平滑曲线推断伪时间变量。主要步骤包括:全局谱系识别、平滑曲线构建、上游分析(基因过滤、标准化、降维)、下游分析(识别动态基因)。slingshot 提供了 wrapper 函数简化操作,同时支持完全无监督或半监督方式进行谱系结构分析。在下游分析中,可以基于 p 值选取动态基因并绘制热图。此外,slingshot 还能处理大规模数据集,并允许指定起点和终点集群。
slingshot 是一个用于单细胞转录组数据轨迹推断的R包,通过构建最小生成树(MST)识别全局谱系结构,并通过平滑曲线推断伪时间变量。主要步骤包括:全局谱系识别、平滑曲线构建、上游分析(基因过滤、标准化、降维)、下游分析(识别动态基因)。slingshot 提供了 wrapper 函数简化操作,同时支持完全无监督或半监督方式进行谱系结构分析。在下游分析中,可以基于 p 值选取动态基因并绘制热图。此外,slingshot 还能处理大规模数据集,并允许指定起点和终点集群。
condiments Paper • condimentsPaper
First example: TGF-beta dataset • bioc2021trajectories
slingshot requires a minimum of two inputs: (1)a matrix representing the cells in a reduced-dimensional space. (2)a vector of cluster labels.
主要步骤:
(1)getLineages function to identify global lineage structrue by construsting MST on the clusters
(2)getCurves function to construct smooth curves and infer the psudotime variable
- simulated dataset 1 (single trajectory) and dataset 2 (bifurcating)
#####dataset1
# generate synthetic count data representing a single lineage
means <- rbind(
# non-DE genes
matrix(rep(rep(c(0.1,0.5,1,2,3), each = 300),100),
ncol = 300, byrow = TRUE),
# early deactivation
matrix(rep(exp(atan( ((300:1)-200)/50 )),50), ncol = 300, byrow = TRUE),
# late deactivation
matrix(rep(exp(atan( ((300:1)-100)/50 )),50), ncol = 300, byrow = TRUE),
# early activation
matrix(rep(exp(atan( ((1:300)-100)/50 )),50), ncol = 300, byrow = TRUE),
# late activation
matrix(rep(exp(atan( ((1:300)-200)/50 )),50), ncol = 300, byrow = TRUE),
# transient
matrix(rep(exp(atan( c((1:100)/33, rep(3,100), (100:1)/33) )),50),
ncol = 300, byrow = TRUE)
)
counts <- apply(means,2,function(cell_means){
total <- rnbinom(1, mu = 7500, size = 4)
rmultinom(1, total, cell_means)
})
rownames(counts) <- paste0('G',1:750)
colnames(counts) <- paste0('c',1:300)
sce <- SingleCellExperiment(assays = List(counts = counts))
#####dataset2
library(slingshot, quietly = FALSE)
data("slingshotExample")
rd <- slingshotExample$rd
cl <- slingshotExample$cl
dim(rd) # data representing cells in a reduced dimensional space
- upstream analysis
(1)gene filtering
# filter genes down to potential cell-type markers
# at least M (15) reads in at least N (15) cells
geneFilter <- apply(assays(sce)$counts,1,function(x){
sum(x >= 3) >= 10
})
sce <- sce[geneFilter, ](2)normalization
normalization可以选择不同方法,并根据不同方法选择noemalization在pipeline里的顺序,例如MNN是先降维在去除batch effect.作者推荐用scone package,模拟数据用quantile normalization.
FQnorm <- function(counts){
rk <- apply(counts,2,rank,ties.method='min')
counts.sort <- apply(counts,2,sort)
refdist <- apply(counts.sort,1,median)
norm <- apply(rk,2,function(r){ refdist[r] })
rownames(norm) <- rownames(counts)
return(norm)
}
assays(sce)$norm <- FQnorm(assays(sce)$counts)(3)降维
示例两种方法:PCA, UMAP
pca <- prcomp(t(log1p(assays(sce)$norm)), scale. = FALSE)
rd1 <- pca$x[,1:2]
plot(rd1, col = rgb(0,0,0,.5), pch=16, asp = 1)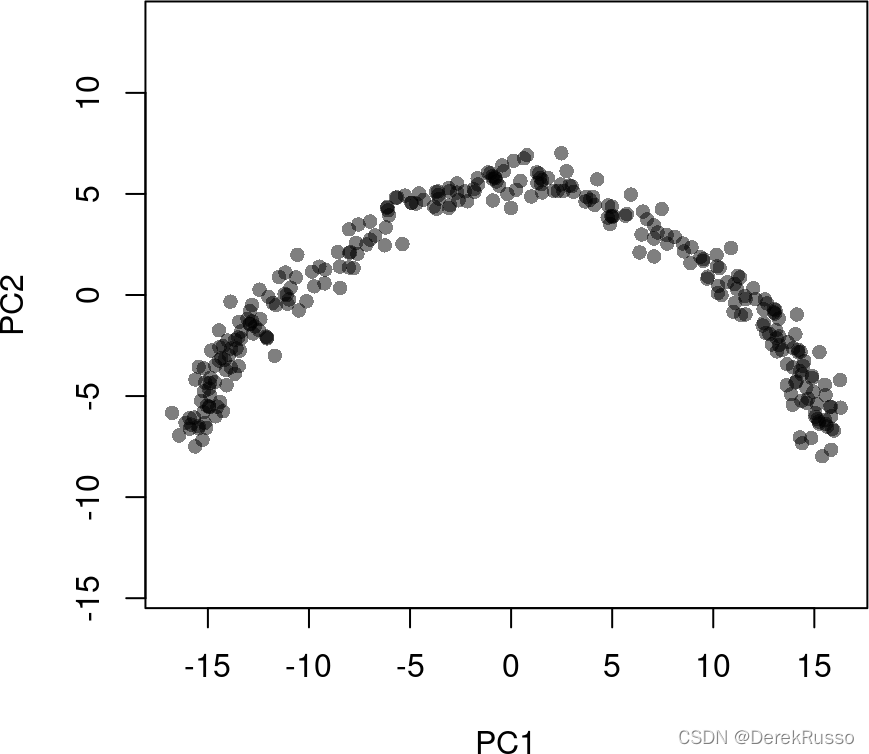
library(uwot)
rd2 <- uwot::umap(t(log1p(assays(sce)$norm)))
colnames(rd2) <- c('UMAP1', 'UMAP2')
plot(rd2, col = rgb(0,0,0,.5), pch=16, asp = 1) 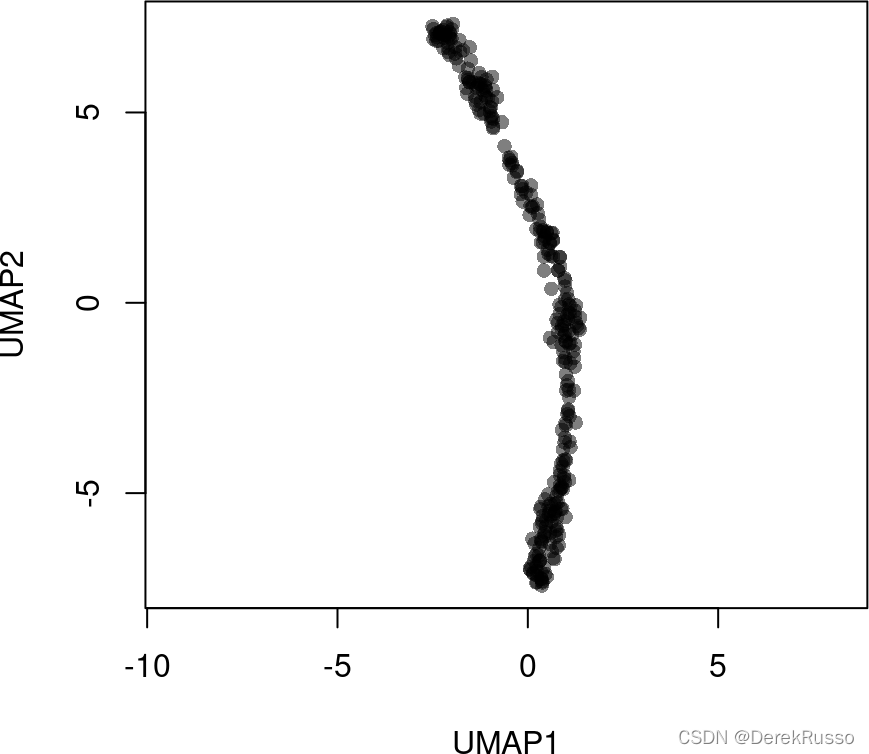
##把两种降维结果都加进sce object,但后续分析用pca结果
reducedDims(sce) <- SimpleList(PCA = rd1, UMAP = rd2)
(3)clustering
两种方法:gaussian mixture modeling (示例) or k-means
library(mclust, quietly = TRUE)
cl1 <- Mclust(rd1)$classification
colData(sce)$GMM <- cl1
library(RColorBrewer)
plot(rd1, col = brewer.pal(9,"Set1")[cl1], pch=16, asp = 1)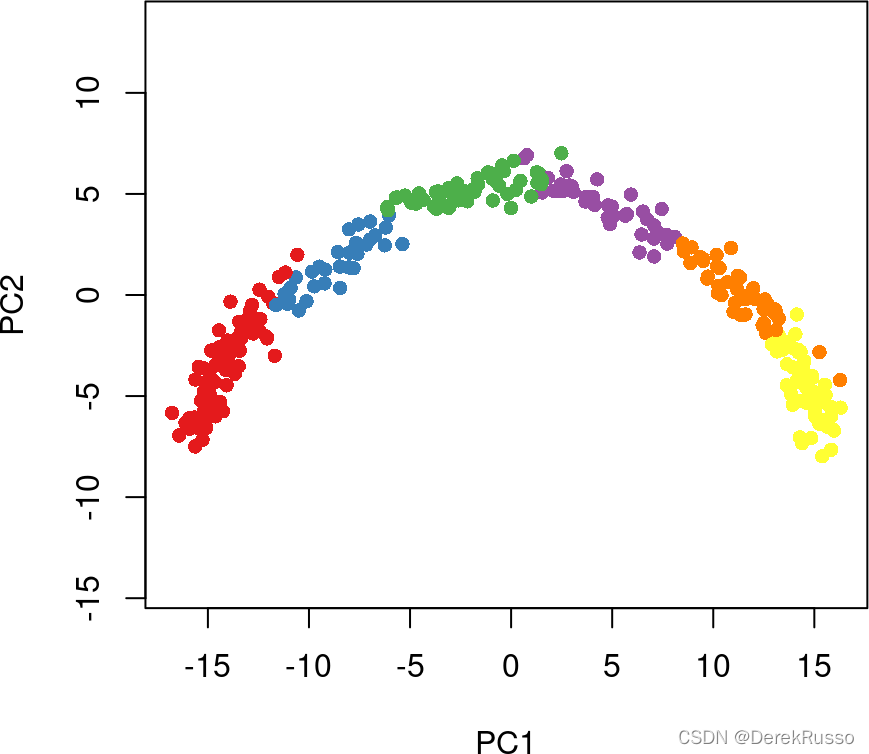
- using slingshot
本质上是两部,第一步发现global structure,第二步fitting simultaneous principal curves.既可以用wrapper function "slingshot" 一句命令就搞定(作者推荐);也可以先后用“getLineages”和“getCurves”两句命令。下面单轨迹dataset用wrapper命令,双轨迹dataset用后两个命令分步完成。
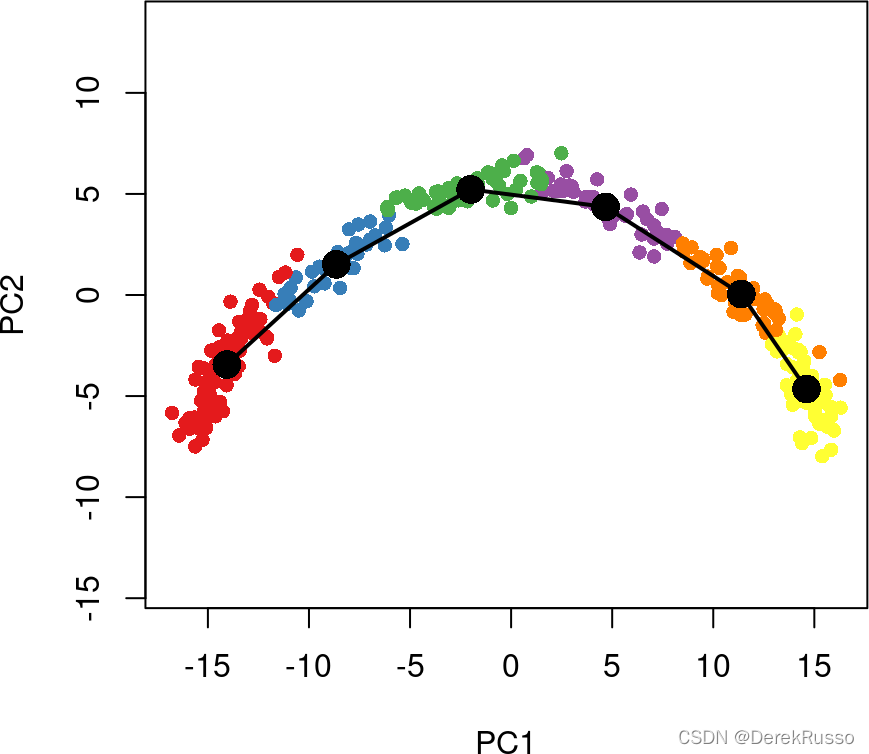
sce <- slingshot(sce, clusterLabels = 'GMM', reducedDim = 'PCA')visualize the result
summary(sce$slingPseudotime_1)
library(grDevices)
colors <- colorRampPalette(brewer.pal(11,'Spectral')[-6])(100)
plotcol <- colors[cut(sce$slingPseudotime_1, breaks=100)]
plot(reducedDims(sce)$PCA, col = plotcol, pch=16, asp = 1)
lines(SlingshotDataSet(sce), lwd=2, col='black')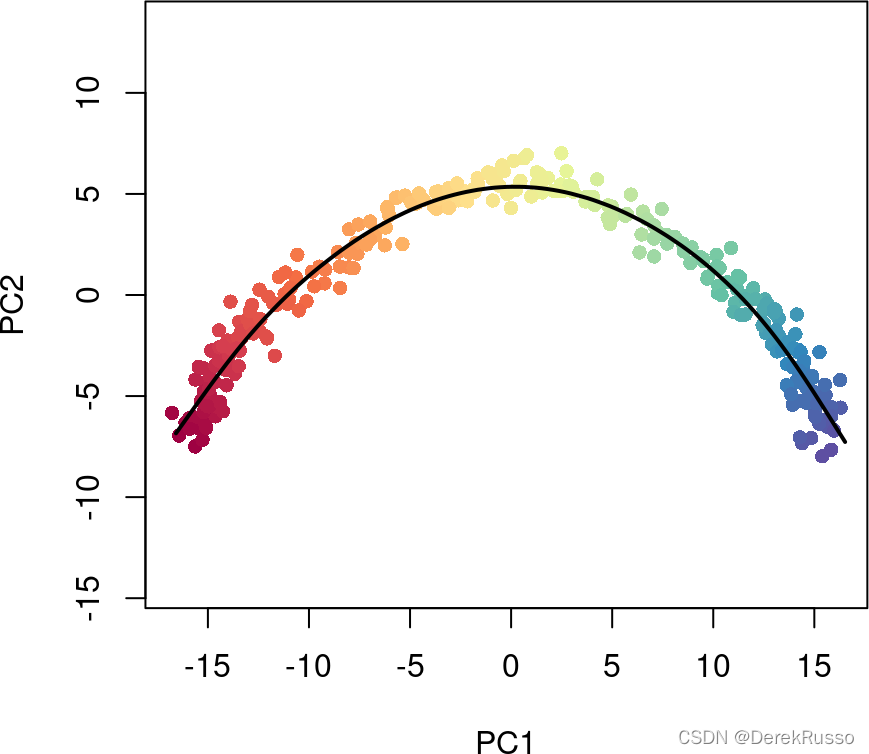
#We can also see how the lineage structure was intially estimated by the cluster-based minimum spanning tree
plot(reducedDims(sce)$PCA, col = brewer.pal(9,'Set1')[sce$GMM], pch=16, asp = 1)
lines(SlingshotDataSet(sce), lwd=2, type = 'lineages', col = 'black')
- downstream analysis (identifying temporally dynamic genes)
library(tradeSeq)
# fit negative binomial GAM
sce <- fitGAM(sce)
# test for dynamic expression
ATres <- associationTest(sce)We can then pick out the top genes based on p-values and visualize their expression over developmental time with a heatmap. Here we use the top 250 most dynamically expressed genes.
topgenes <- rownames(ATres[order(ATres$pvalue), ])[1:250]
pst.ord <- order(sce$slingPseudotime_1, na.last = NA)
heatdata <- assays(sce)$counts[topgenes, pst.ord]
heatclus <- sce$GMM[pst.ord]
heatmap(log1p(heatdata), Colv = NA,
ColSideColors = brewer.pal(9,"Set1")[heatclus]) 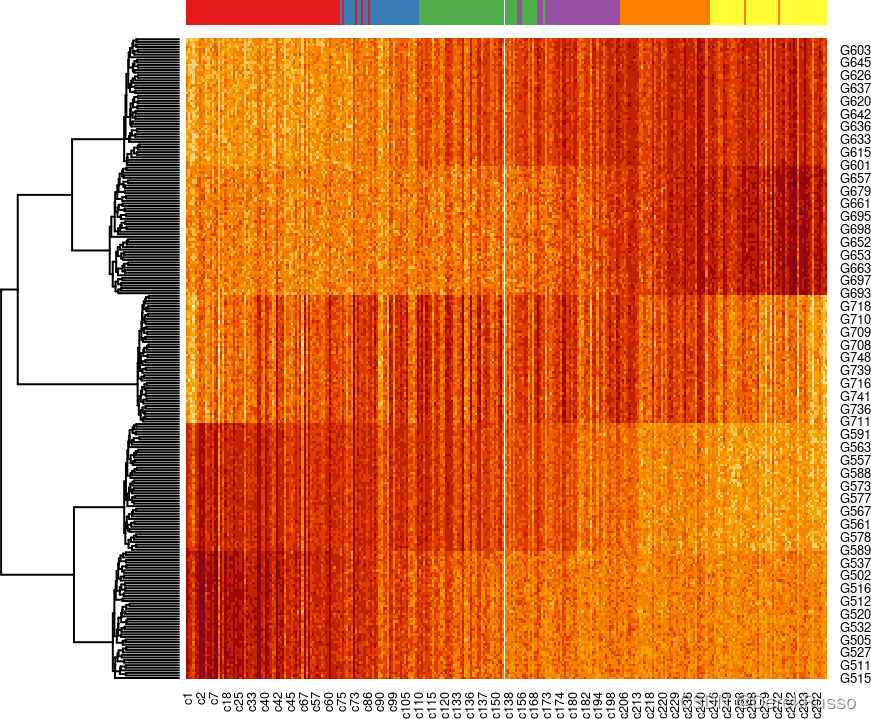
- Detailed slingshot functionalities
为了更好的解释slingshot的其他功能,以下开始用slingshot自带的第二个数据集(bifurcating),并且直接将low dimensional matrix (by k-means)和cluster labels 作为输入,而非先构建sce object.因为后者还需要表达矩阵。
(1)identifying global lineage structure
The getLineages function takes as input an n ×× p matrix and a vector of clustering results of length n.
This analysis can be performed in an entirely unsupervised manner or in a semi-supervised manner by specifying known initial and terminal point clusters.
If we do not specify a starting point, slingshot selects one based on parsimony, maximizing the number of clusters shared between lineages before a split.
we generally recommend the specification of an initial cluster based on prior knowledge (either time of sample collection or established gene markers).
lin1 <- getLineages(rd, cl, start.clus = '1')
lin1
plot(rd, col = brewer.pal(9,"Set1")[cl], asp = 1, pch = 16)
lines(SlingshotDataSet(lin1), lwd = 3, col = 'black')
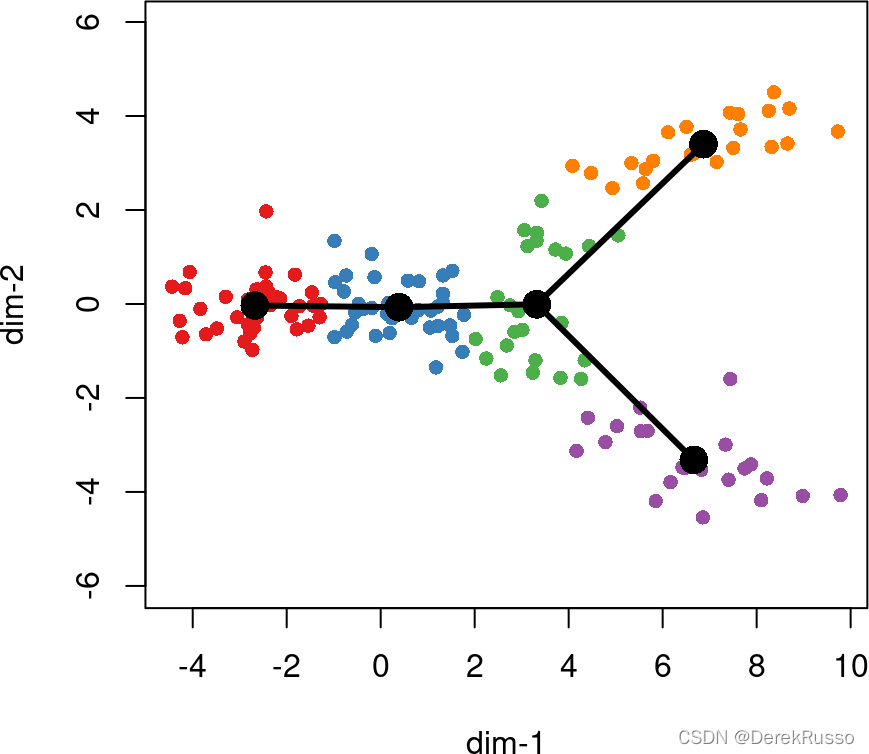
slingshot also allows for the specification of known endpoints. Clusters which are specified as terminal cell states will be constrained to have only one connection when the MST is constructed (ie., they must be leaf nodes). 注意!如果指定了terminal cells,那么它将是唯一终点且只有一个联系。
lin2 <- getLineages(rd, cl, start.clus= '1', end.clus = '3')
plot(rd, col = brewer.pal(9,"Set1")[cl], asp = 1, pch = 16)
lines(SlingshotDataSet(lin2), lwd = 3, col = 'black', show.constraints = TRUE) 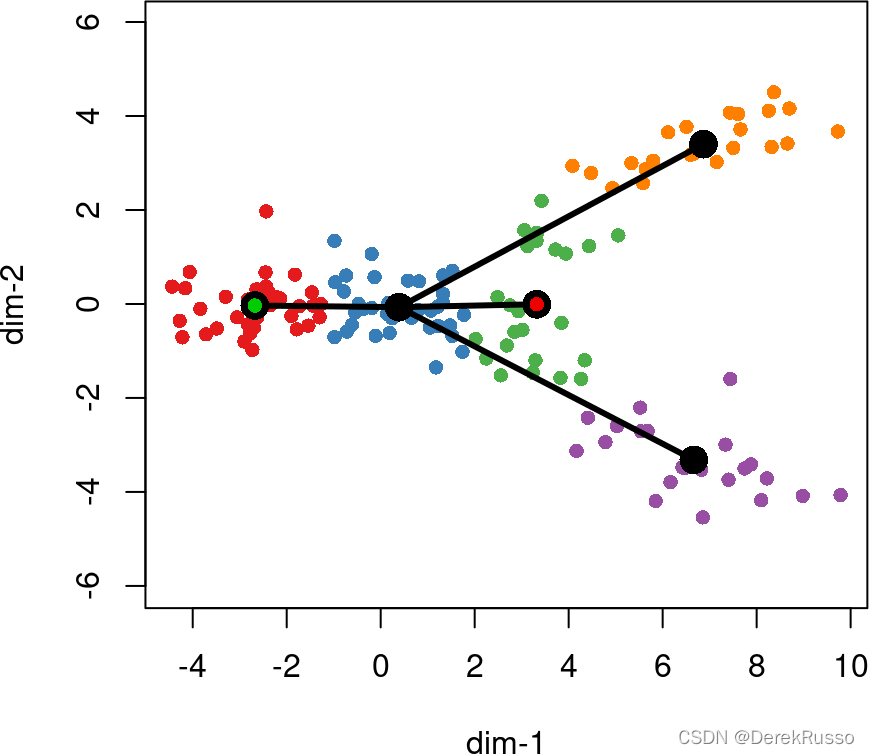
(2)constructing smooth curves and ordering cells
crv1 <- getCurves(lin1)
crv1
plot(rd, col = brewer.pal(9,"Set1")[cl], asp = 1, pch = 16)
lines(SlingshotDataSet(crv1), lwd = 3, col = 'black')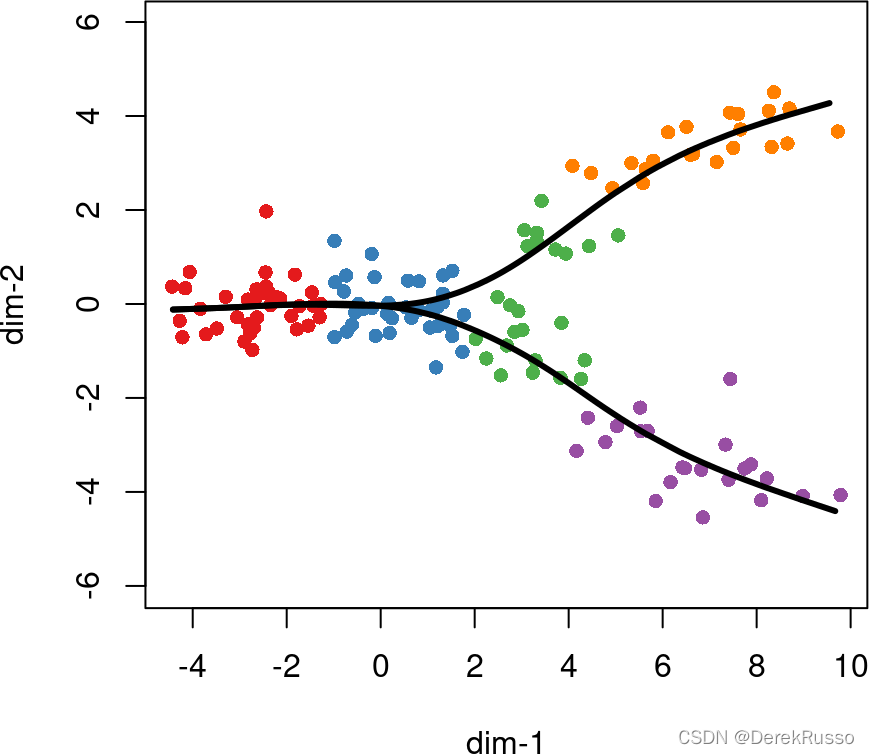
(3)running slingshot on large datasets
在slingshot或者getCurves功能中,可以用approx_points参数去调节projection resolution.官方推荐100-200之间,默认150.如果设置approx_points = F,则有多少细胞便会投影出多少个点,计算量也非常的大。

















 1479
1479

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









