




用fastqc进行数据质控:
Babraham Bioinformatics - Public Projects Download在官网进行下载

fastqc在windows系统下运行完全没问题
把这个文件下载下来
这里主要要配置环境变量:主要是fastqc的和java的
在文件夹中运行cmd
这样输入就讲fastqc打开了

这里用一个名为ERR499.R1.fq的文件演示一下:
<<ERR499.R1.fq>>
直接导入fastqc:
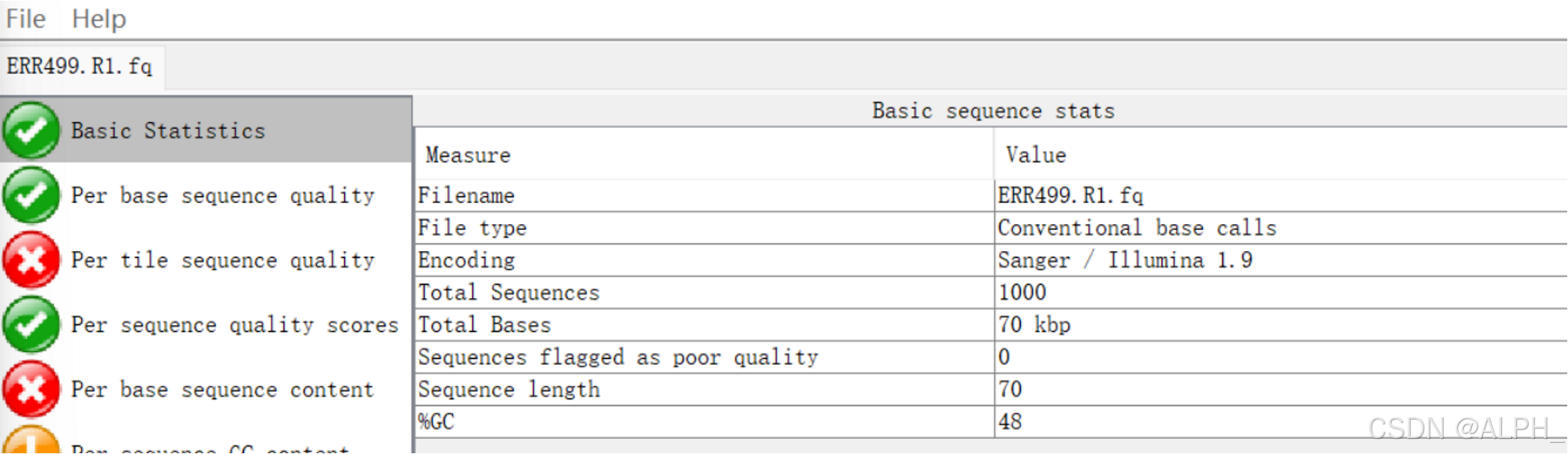
下面来解析结果:
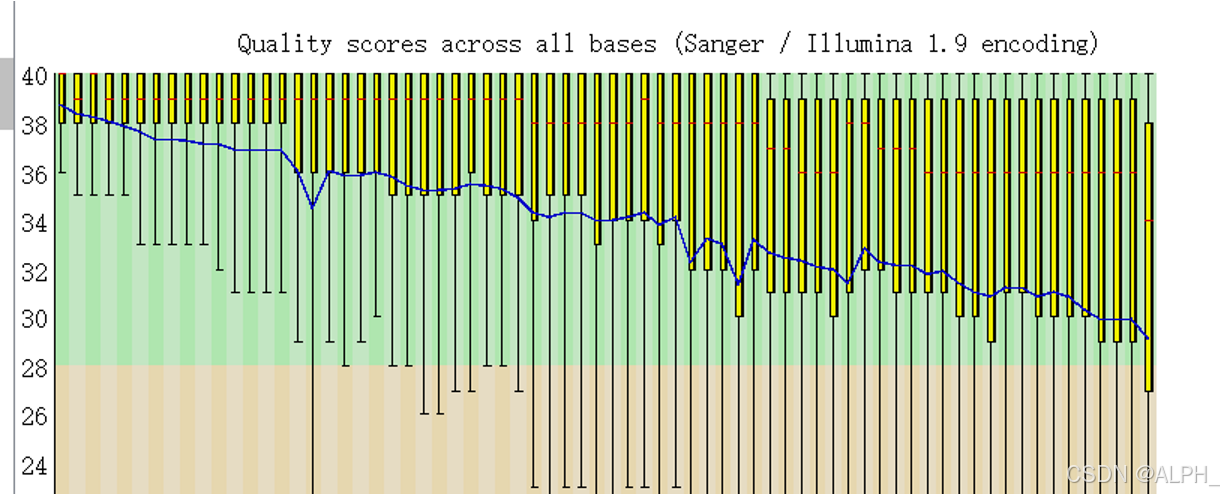
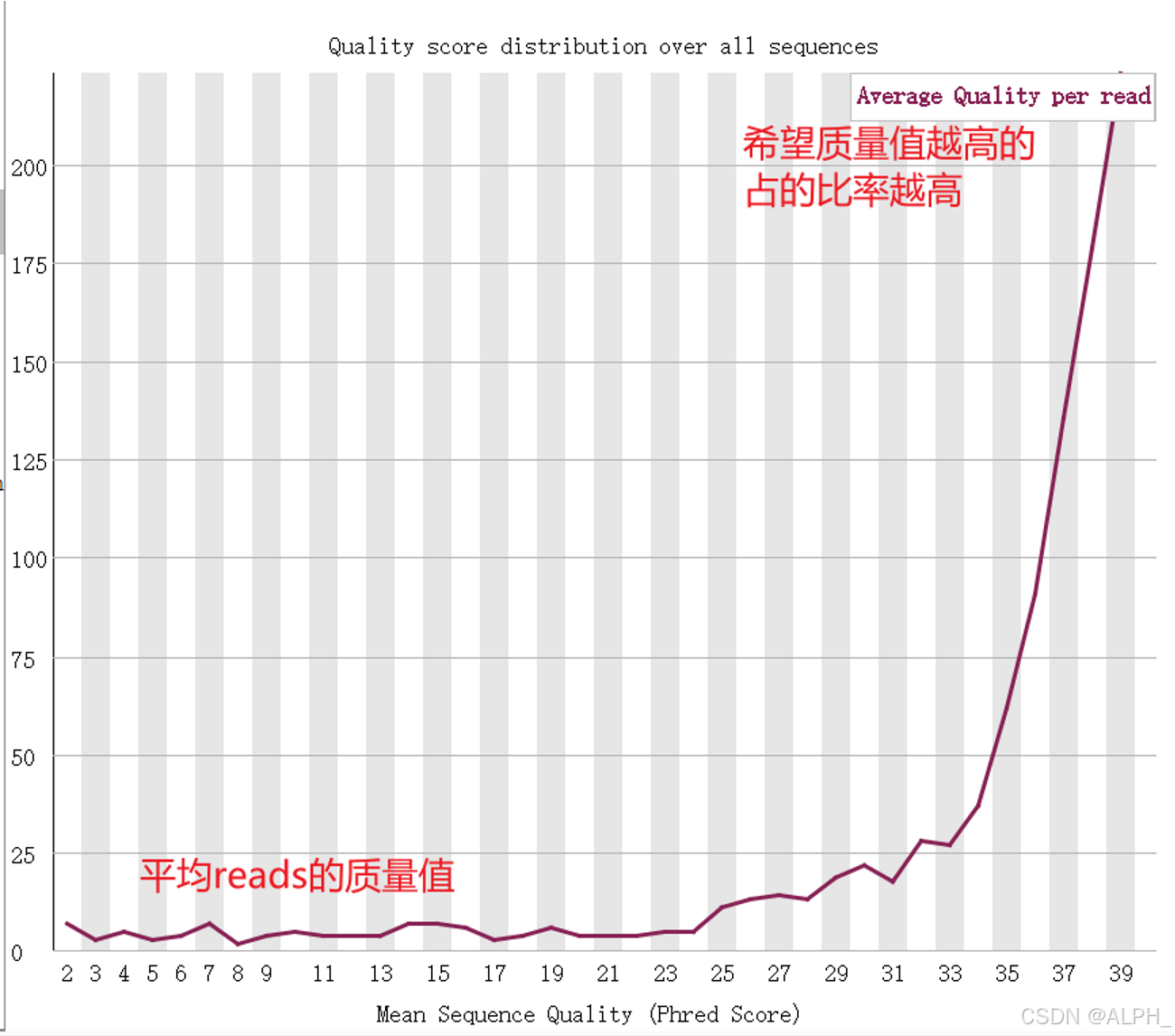
整体在28以上,还可以,如果在红区的太多就是不好的
蓝色就是好的,有其他颜色就是不好的
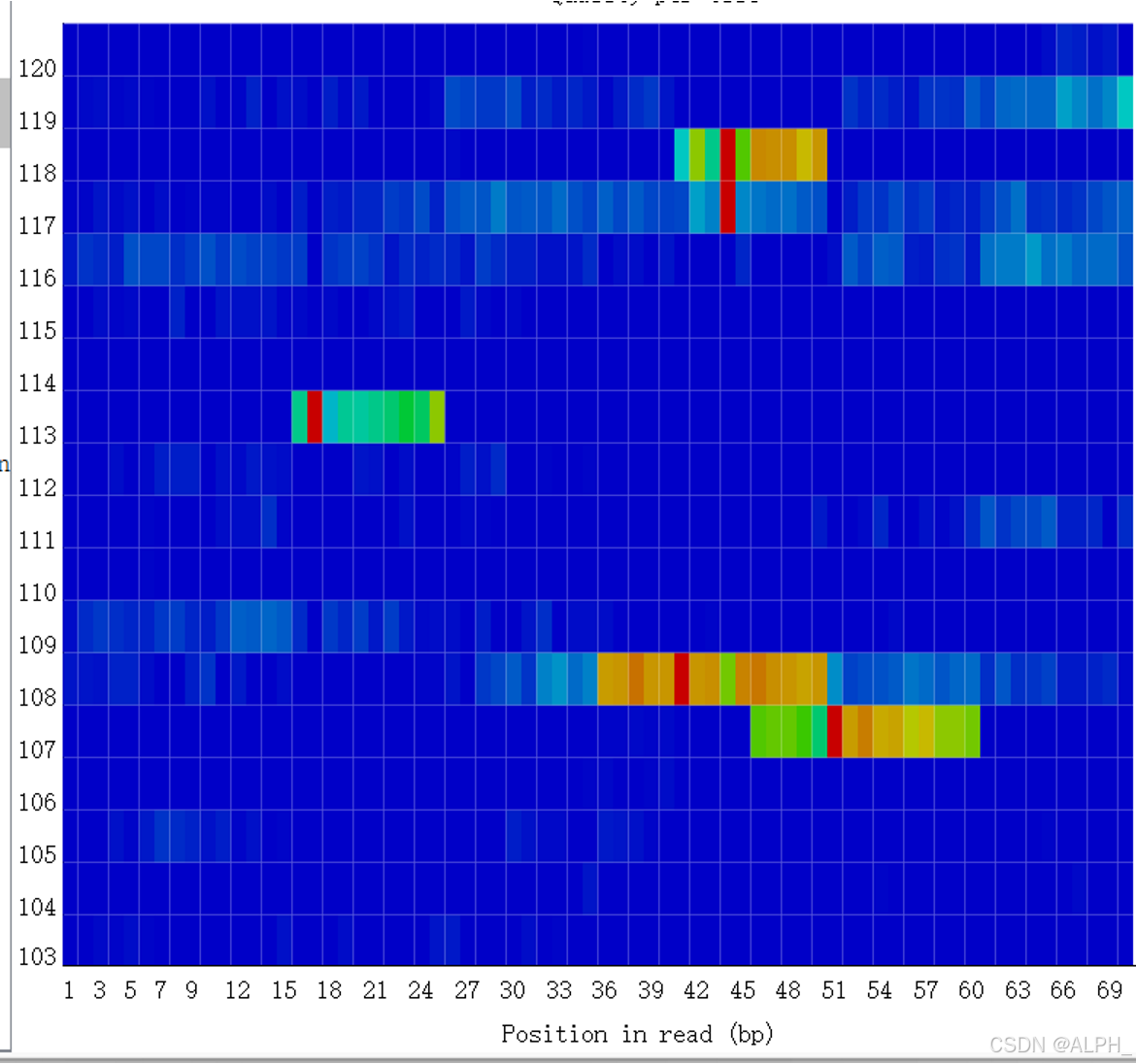
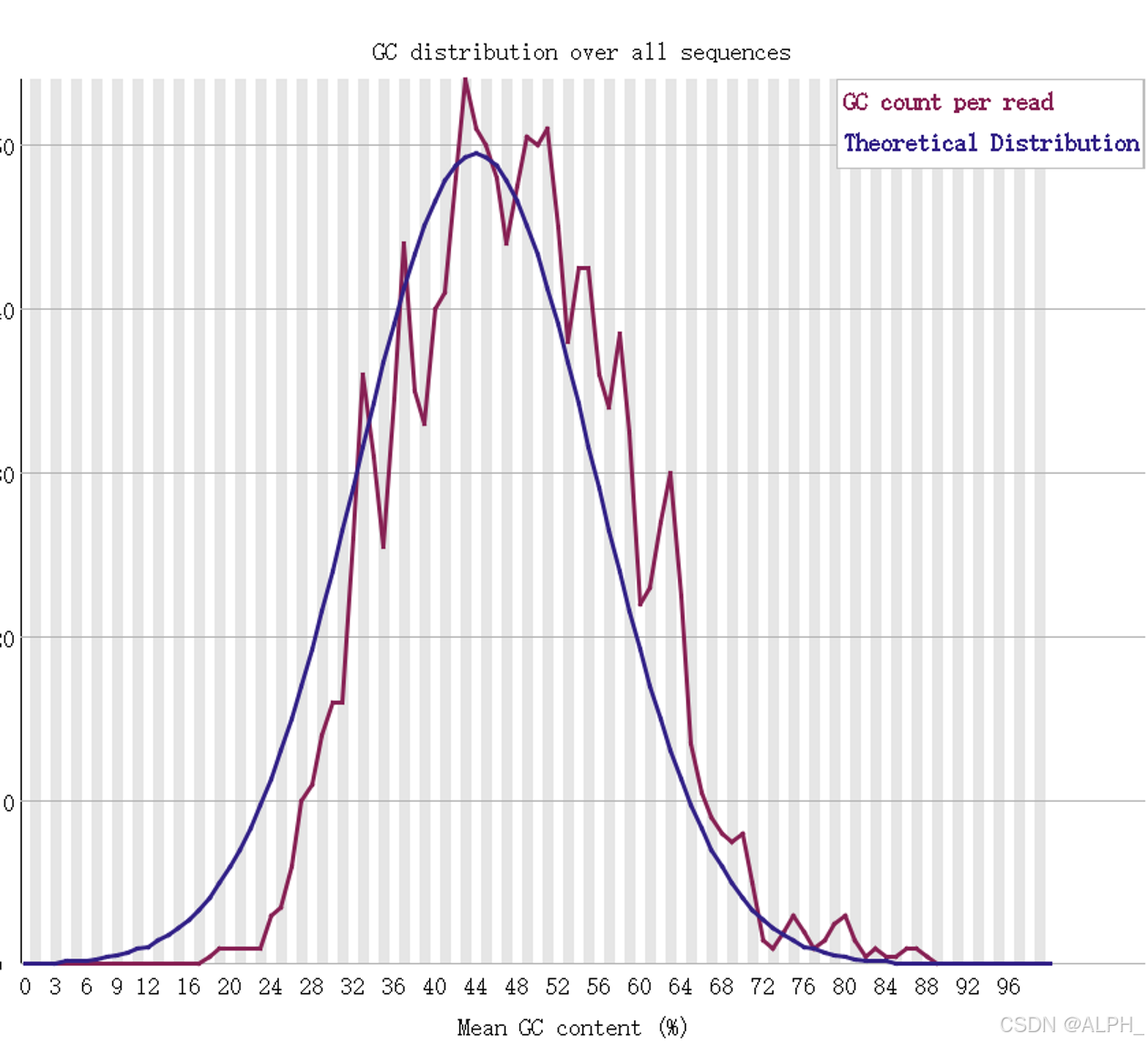
是单峰,不是双峰
人一般是49%
如果出现双峰:
1.就说明可能测到了杂质物种的峰值
2.片段打断不均匀
下面我们要去除adapt,这里安装cutadapt:
为什么要去除adaptor:
一般来说,测序公司会提供相关的适配子序列信息。如果你自己从网上下载数据,通常可以根据文章中的描述来查找这些信息。比如,文章里可能会提到适配子序列是否包含在内,或者可以查看该测序实验的建库类型。如果是Illumina平台进行的测序,我们就可以查找Illumina的出塞序列(例如,I7或I5的引物序列),这可以通过Illumina的官方文档或者其他资源找到。
假设现在我们确定了适配子的序列,接下来就要处理适配子去除的问题。比如,在70bp的测序中,如果读长只有40bp或50bp,那么在测序时,读长不足时,可能会测到部分适配子序列。对于Illumina平台的测序,如果你只获得了40bp的序列,这意味着你有30bp的适配子序列是需要去除的。
因为测序结果可能会带有30bp的适配子序列,如果不去除,接下来的比对就无法正确进行,因为最终比对的序列(假设是70bp)会与基因组序列不匹配。所以,我们需要先去除适配子序列(30bp),然后剩下的40bp才能用于正确的比对,得到可靠的结果。
接下来,我们就可以进行去除适配子的计算(通常用工具如cutadapt、Trimmomatic等)。
具体操作:
cutadapt -a AGATCGGAAGAGCACACGTCTGAACTCCAGTC ERR500.R1.fq -m 30 --output=ERR500.cutAd1.fq

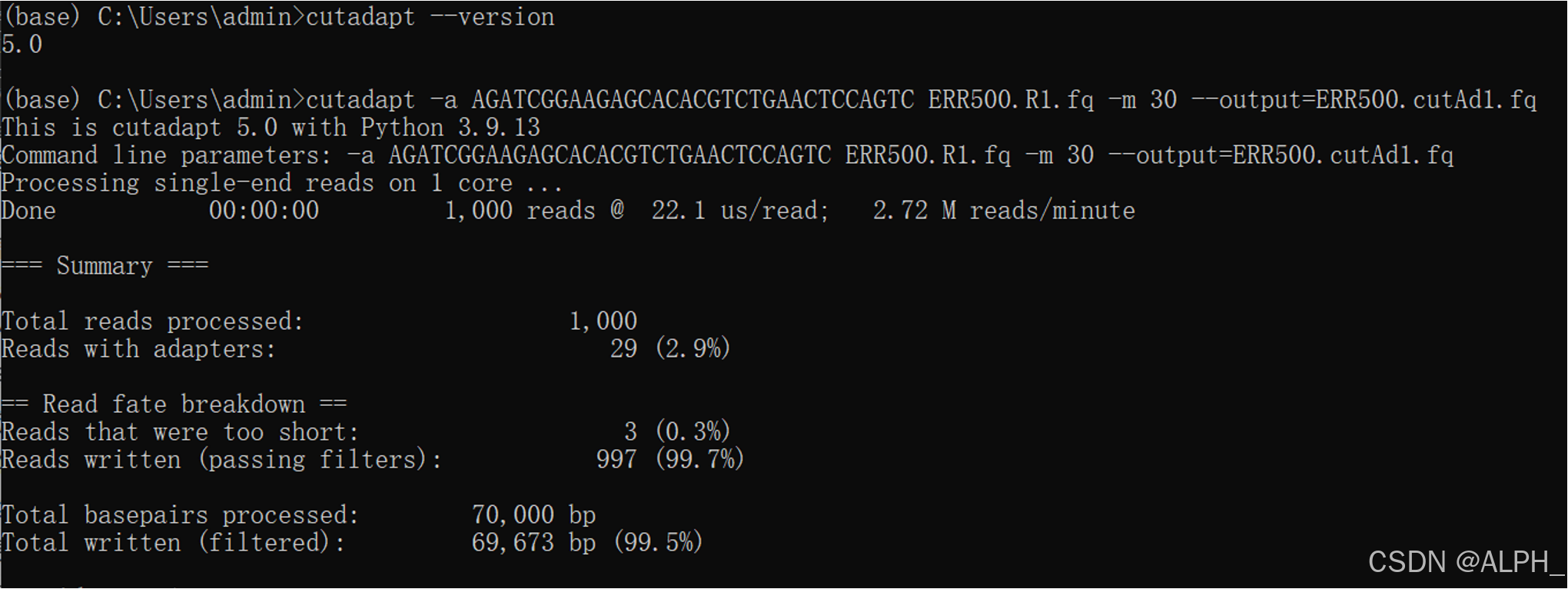
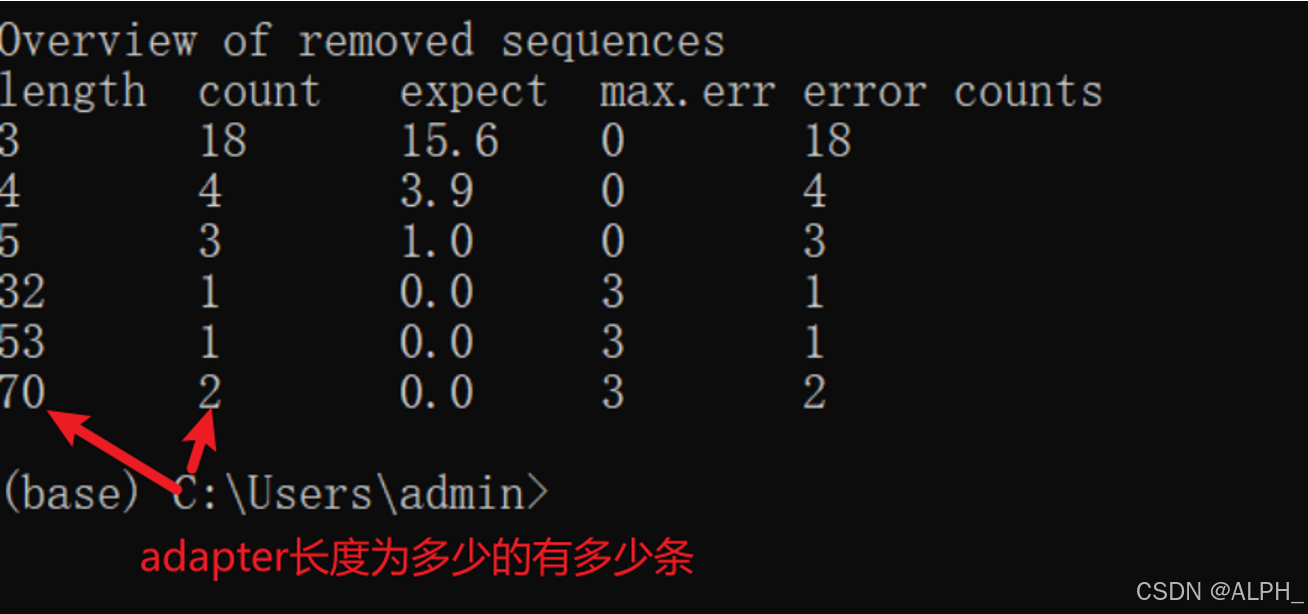
去完adapter后序列长度不一样了
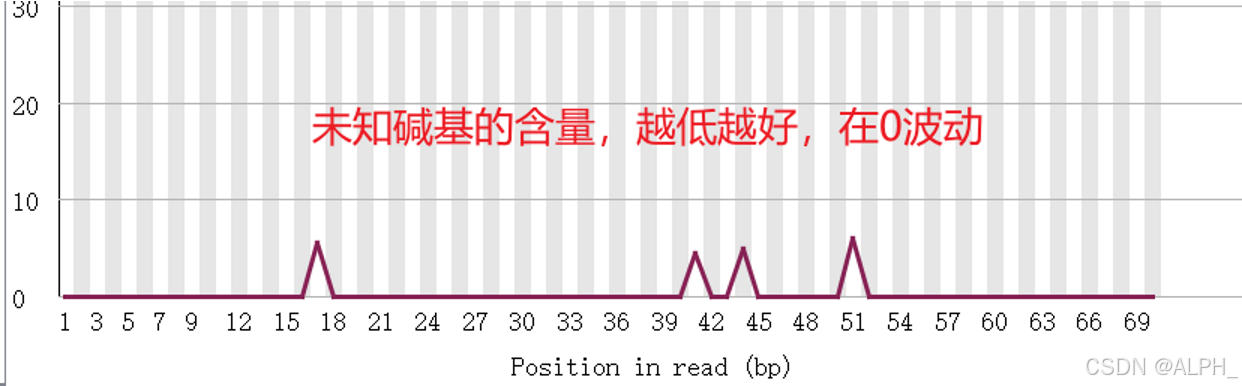
去除N(也就是未知碱基):
read里面都可能会出现n的序列,如果这个n达到了5%或者10%的时候我们就要把它去除掉
这里我
 最低0.47元/天 解锁文章
最低0.47元/天 解锁文章


















 1万+
1万+

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









