



工具链接:https://www.henbio.com/tools
单细胞转录组学火了,可 R 包、Python 脚本、服务器配置……每一步都是劝退级门槛。HiOmics 把 Seurat、SingleR、CellChat 等主流算法搬到云端,十来个模块连成一条“傻瓜式”流水线:上传数据 → 点选参数 → 一键运行 → 直接出图,全程零代码。电脑只要联网,就能在浏览器里完成从质控到文章级可视化。
3 分钟速通全流程
登录与入口
打开 https://www.henbio.com/tools → 云分析流程化 → 单细胞转录组分析。
数据读入 + 质控分群
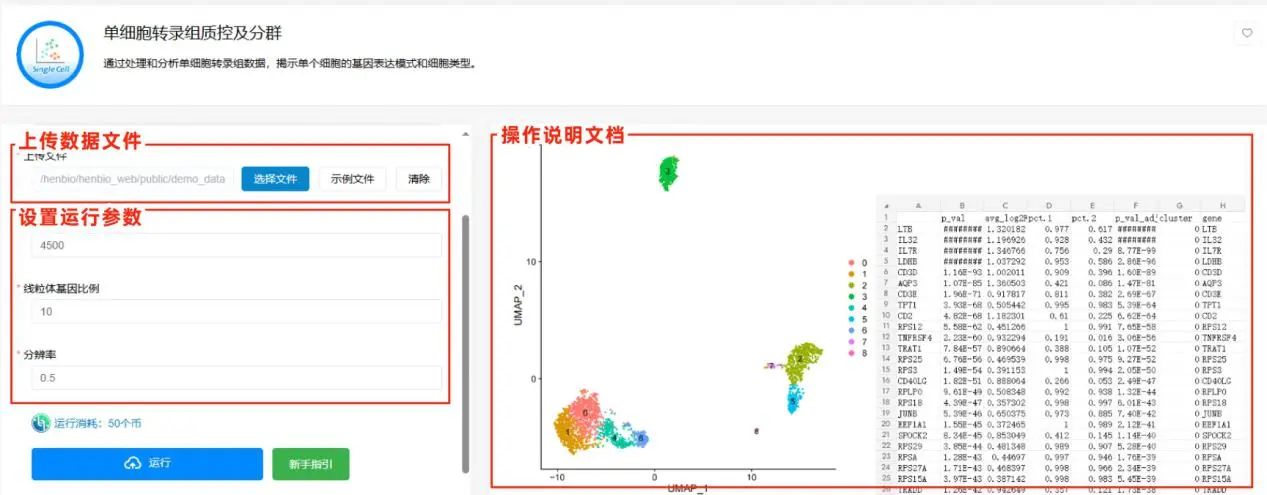
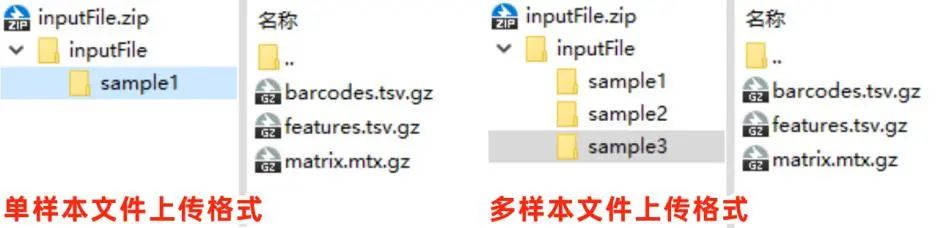
• 上传压缩包:zip 格式,根目录必须是 inputFile;里边放 10X 标准文件夹,此文件夹自行命名(sample1/sample2…)。每个文件夹只对应一个样本。(如下示例所示)
• 设置关键参数:
– 基因数:根据小提琴图nFeature_RNA项,选择合适的基因数进行过滤,离群点判定为质量较差的细胞
– 线粒体比例:选择合适的线粒体基因比例,一般线粒体基因比例过高,则判定为质量较差的细胞(建议不超过20)
– 分辨率:分辨率越大,分群得到的群数越多(建议不超过1),默认为0.5
• 点击【运行】,服务器运算后输出单细胞分群cluster等结果文件,其中的Seuratproject.rds文件要用作为下一步分析的输入数据文件。
细胞类型注释
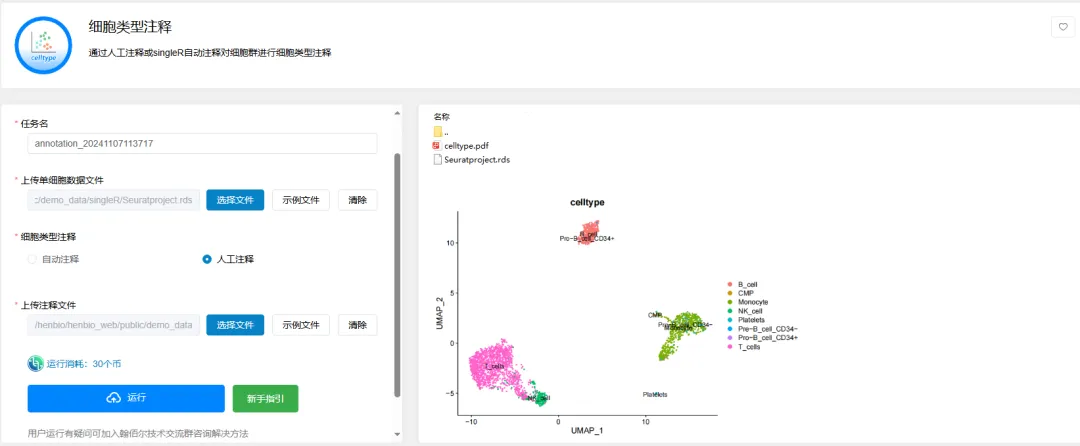
点击【细胞类型注释】→选择【单细胞转录组质控及分群】后得到的Seuratproject.rds文件→通过selfannotation人工方式或singleR自动方式两种方式对第一步的单细胞分群结果进行注释。选择人工方式需上传对应注释文件,选择自动方式需选择参考数据集→点击【运行】
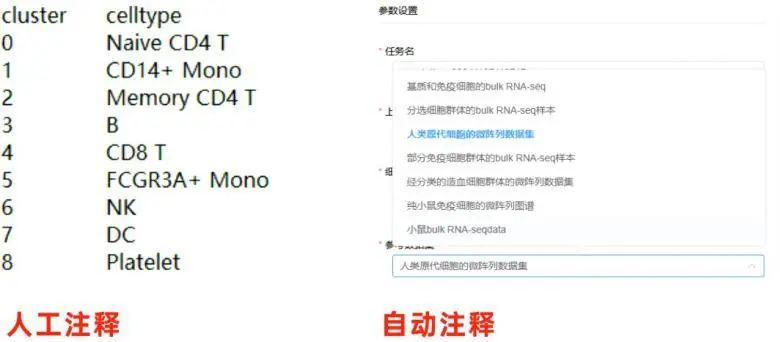
运行得到的结果为细胞分群降维图和Seuratproject.rds文件,依据此文件就可以开展后面的大部分的单细胞分析了。
单细胞转录组构成比
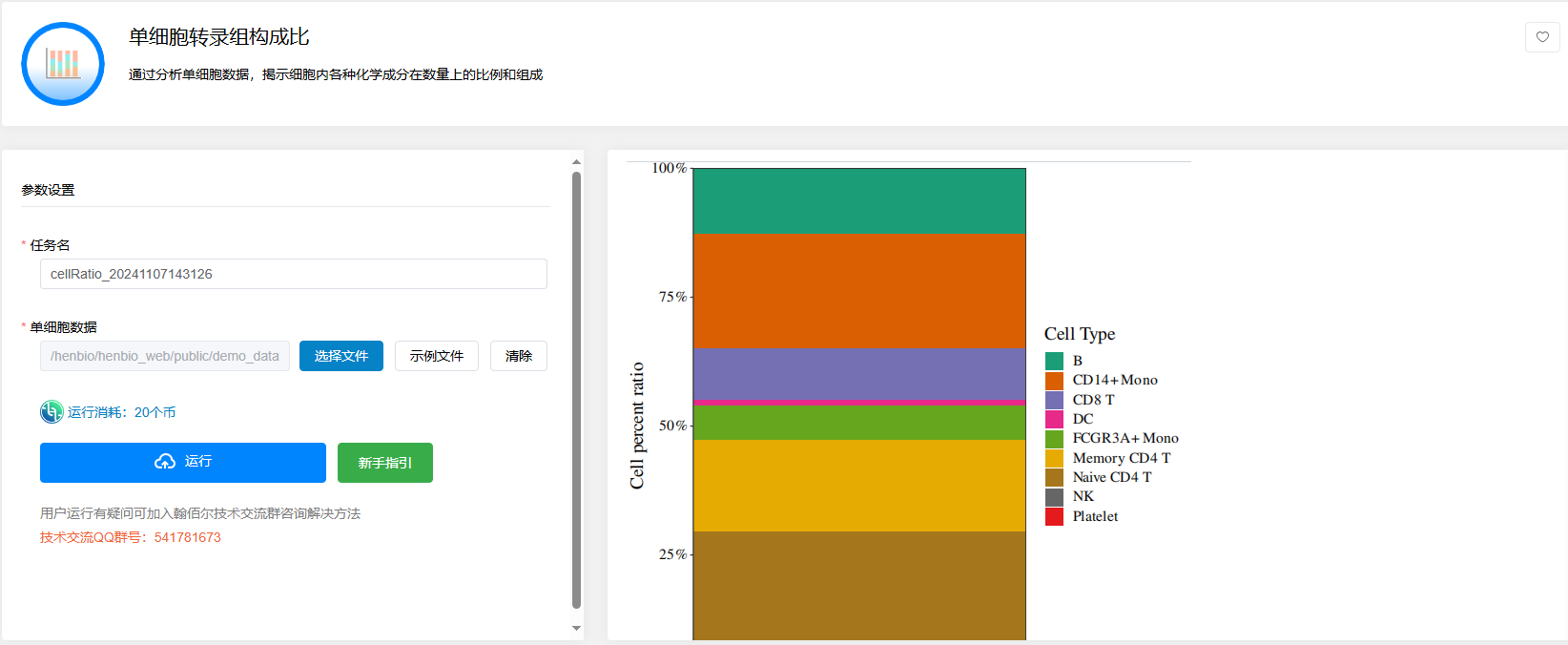
点击【单细胞转录组构成比】→选择【细胞类型注释】后得到的Seuratproject.rds文件→点击【运行】
通过分析单细胞数据,揭示细胞内各种化学成分在数量上的比例和组成。
单细胞表达谱矩阵的获取
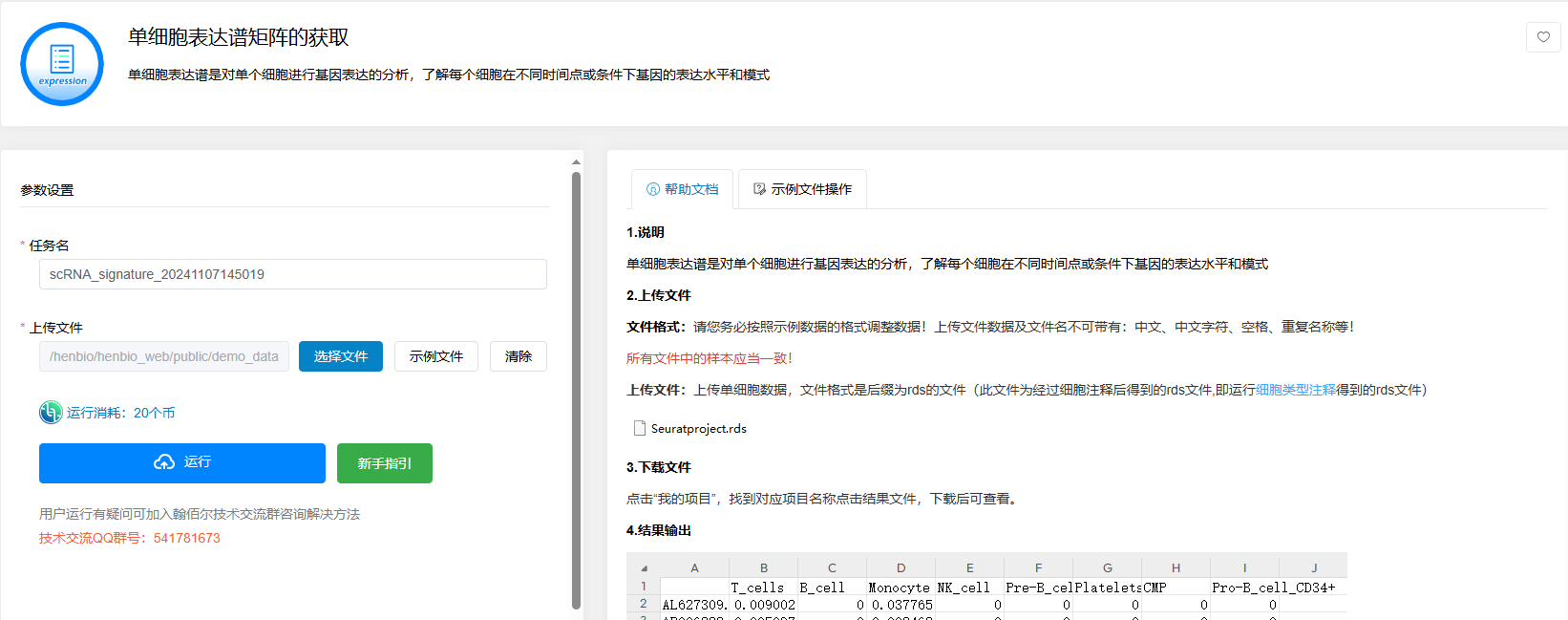
点击【单细胞表达谱矩阵的获取】→选择【细胞类型注释】后得到的Seuratproject.rds文件→点击【运行】
单细胞表达谱是对单个细胞进行基因表达的分析,了解每个细胞在不同时间点或条件下基因的表达水平和模式。
单细胞基因表达量密度图
点击【单细胞基因表达量密度图】→选择【细胞类型注释】后得到的Seuratproject.rds文件→选择【基因文件】→点击【运行】
密度图可以展示不同基因在单细胞中的表达水平分布情况,帮助研究者快速了解哪些基因在细胞群体中表达较高或较低。
细胞通讯分析(一)
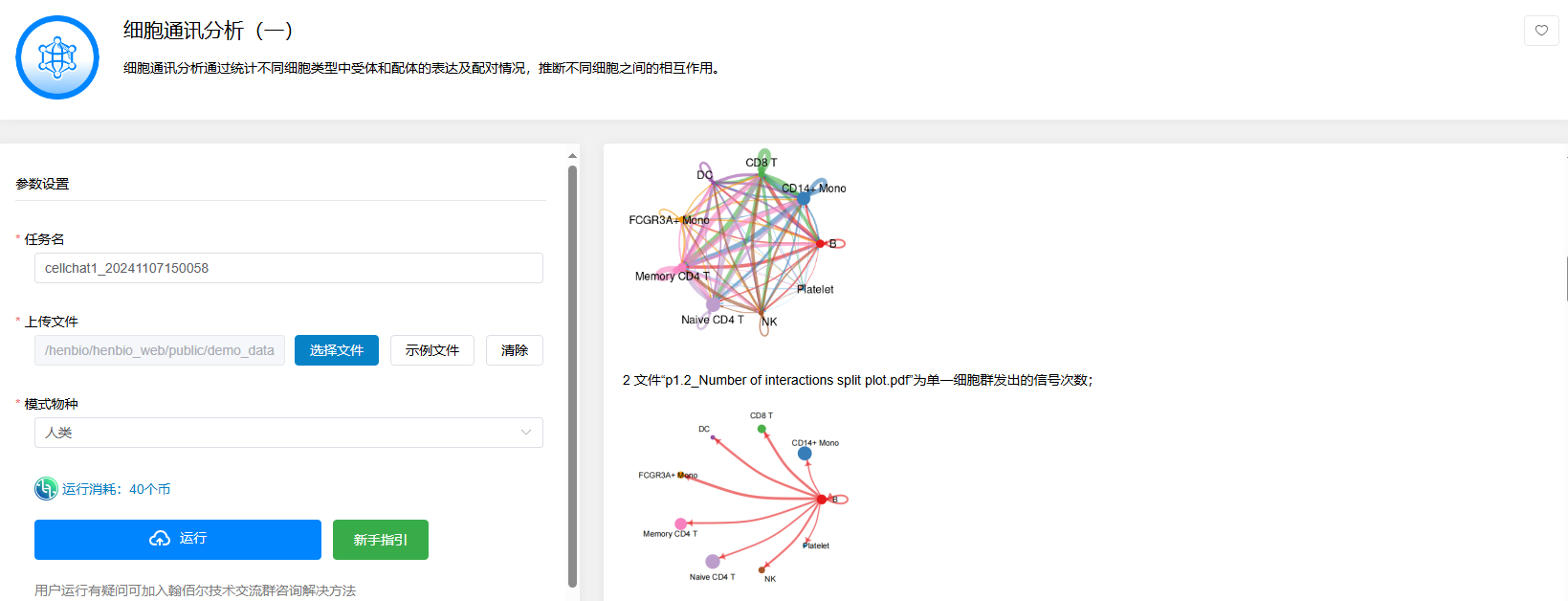
点击【细胞通讯分析(一)】→选择【细胞类型注释】后得到的Seuratproject.rds文件→选择【物种模式】→点击【运行】
细胞通讯分析(二)
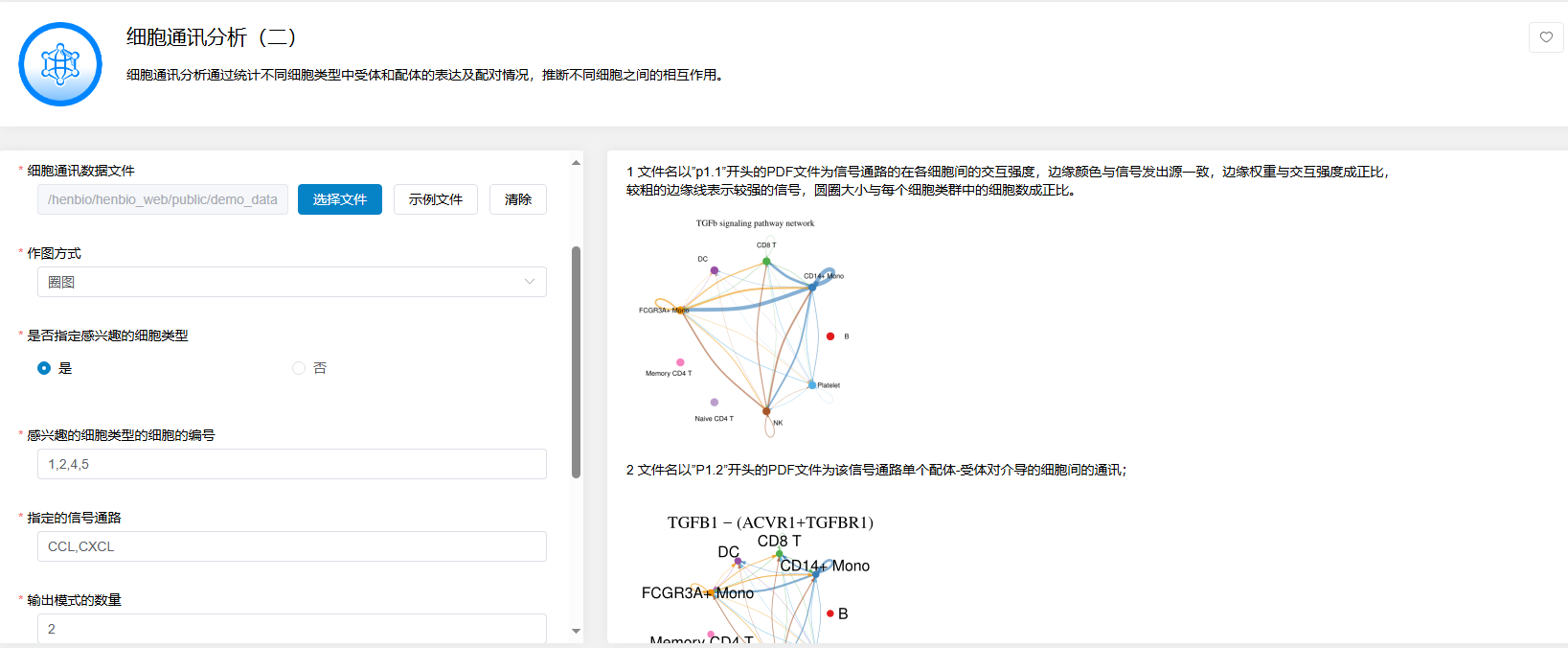
点击【细胞通讯分析(二)】→选择【细胞通讯分析(一)】后得到的Seuratproject.rds文件→选择【作图方式】→选择【是否指定感兴趣的细胞类型】等设置→点击【运行】
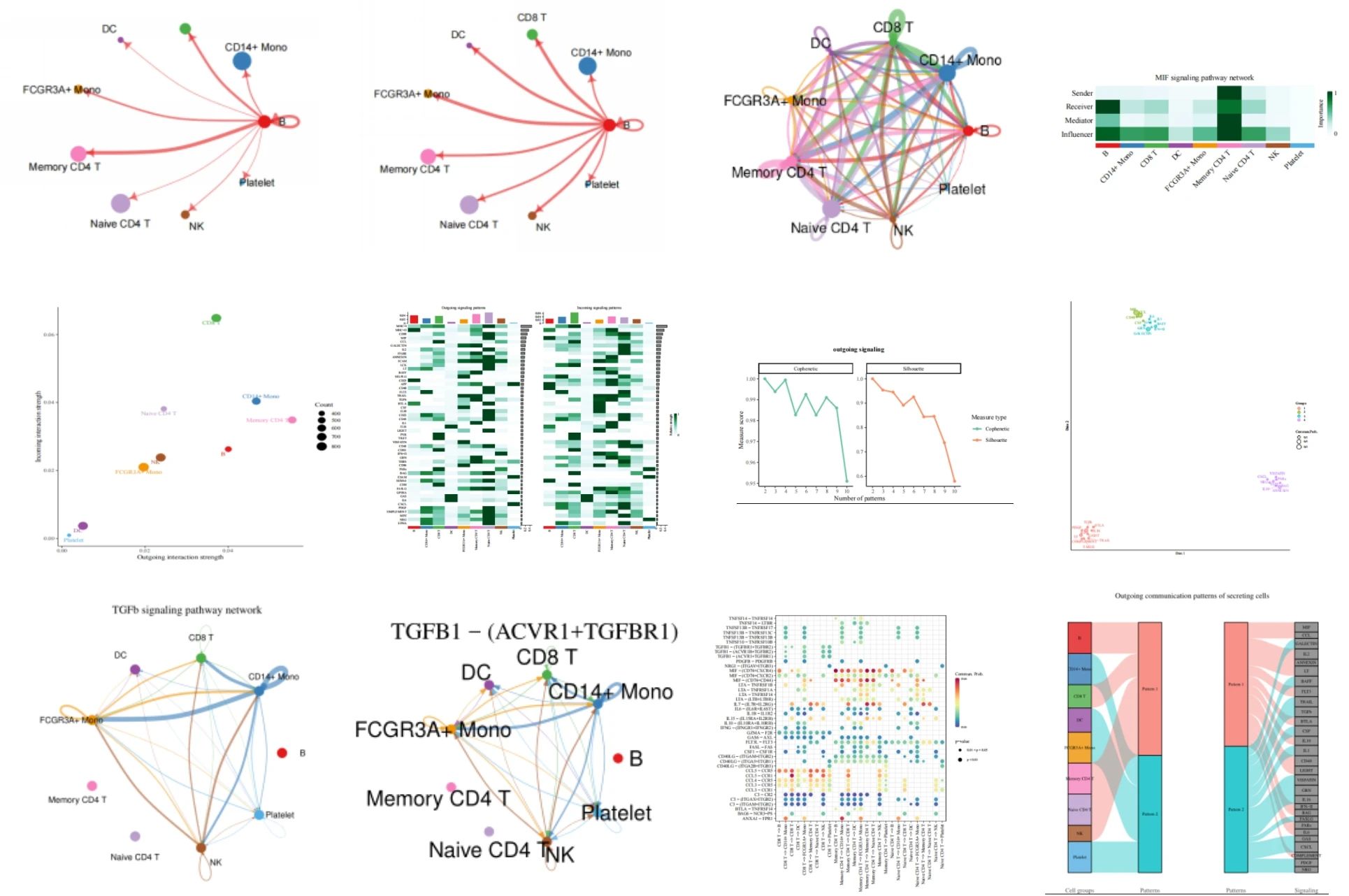
细胞通讯分析通过统计不同细胞类型中受体和配体的表达及配对情况,推断不同细胞之间的相互作用。
单细胞基因表达堆叠小提琴图
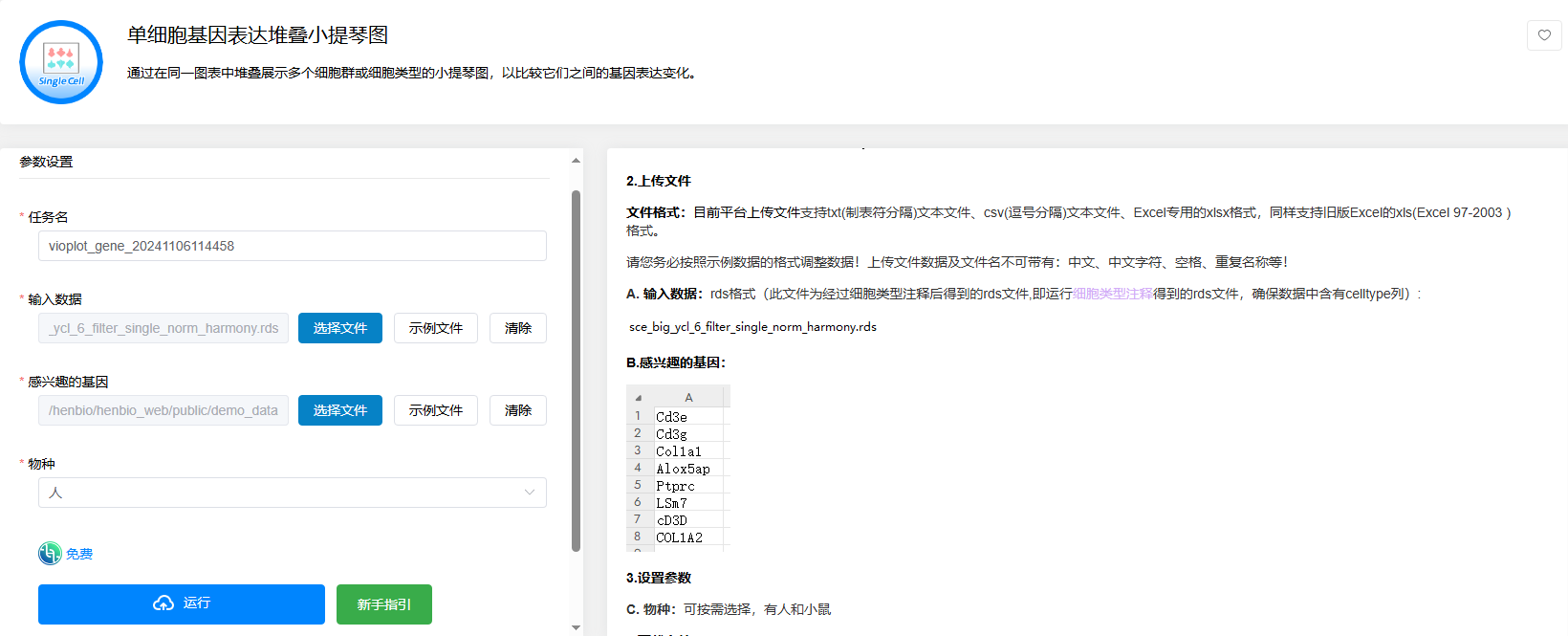
点击【单细胞基因表达堆叠小提琴图】→选择【细胞类型注释】后得到的Seuratproject.rds文件→选择【感兴趣的基因】文件→选择【物种】→点击【运行】
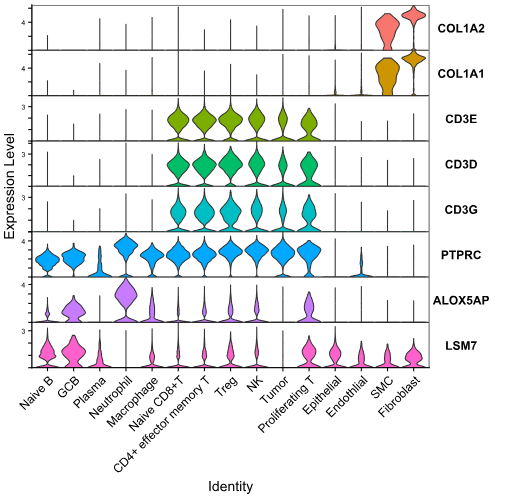
通过在同一图表中堆叠展示多个细胞群或细胞类型的小提琴图,以比较它们之间的基因表达变化。
单细胞富集分析(KEGG)
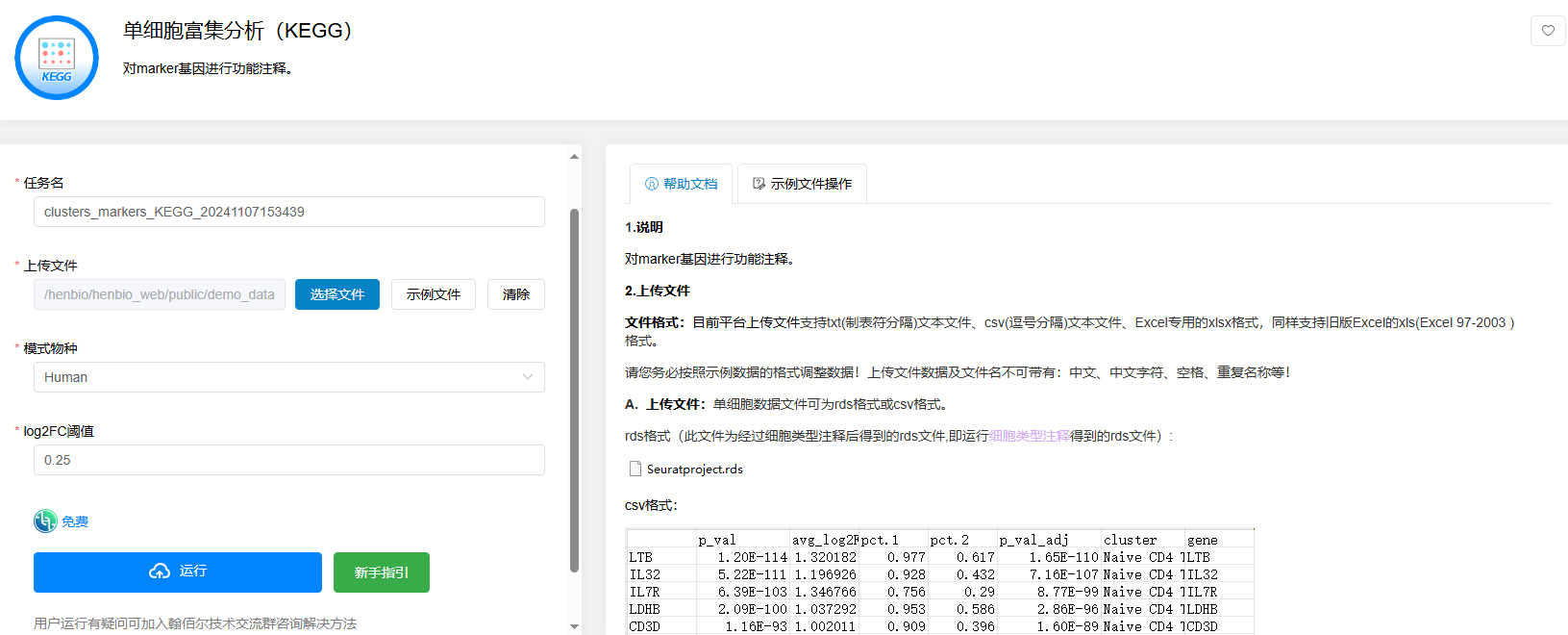
点击【单细胞富集分析(KEGG)】→选择【细胞类型注释】后得到的Seuratproject.rds文件→选择【模式物种】文件→选择【log2FC阈值】→点击【运行】
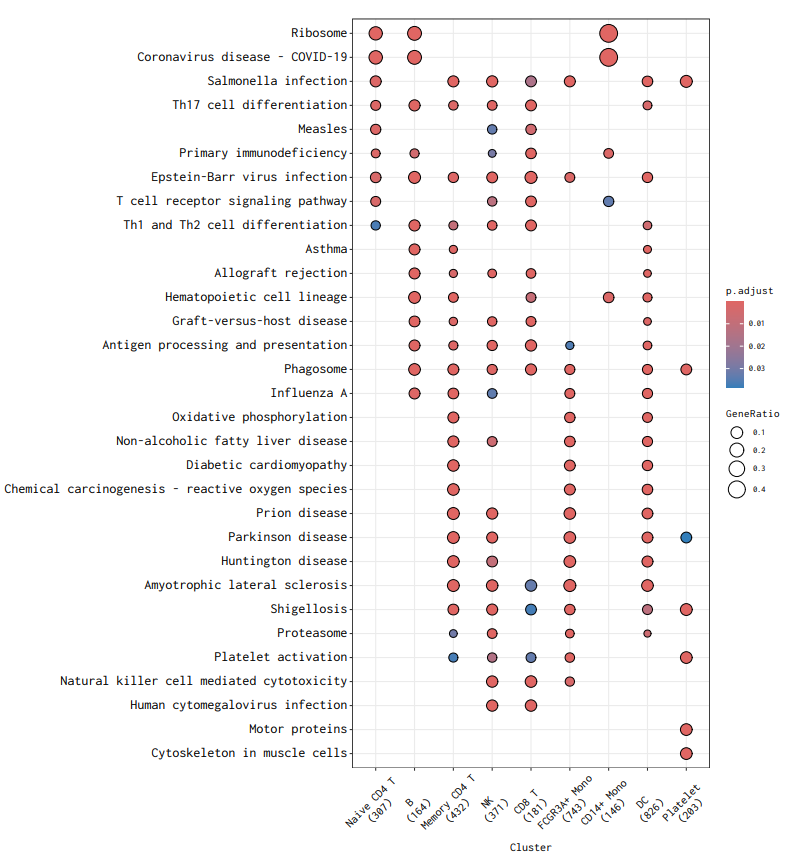
KEGG富集分析揭示单细胞生物学功能、通路和疾病关联
当单细胞技术从“前沿”走向“标配”,我们需要的不再是“会写代码的人”,而是“会提科学问题的人”。HiOmics 把底层技术封装成一键式体验,让研究者把时间和创造力留给真正的生物学发现。
如果您觉得这个网站对您有帮助,还请您帮忙多多转发,与他人分享,让更多人受益。如果内容有任何侵权或是错误,恳请您及时联系我,我一定第一时间改正,感谢!

















 2218
2218

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









