




引言
单细胞RNA测序(scRNA-seq)技术的出现彻底改变了我们研究复杂生物系统的方式,使科学家能够在前所未有的精细水平上解析细胞异质性。传统的bulk RNA测序只能捕获细胞群体的平均表达特征,而单细胞转录组测序允许我们检测每个细胞的基因表达谱,从而揭示被平均效应掩盖的关键生物学信息。本文将全面介绍单细胞RNA测序数据分析的完整流程,重点展示`scrna_toolkit`包在简化分析过程和增强数据可视化方面的应用。
第一部分:单细胞RNA测序技术原理
单细胞RNA测序技术经历了从早期的手动分离单细胞到现代高通量微流控和微滴技术的快速发展。目前主流的技术平台包括10x Genomics Chromium、Drop-seq和Smart-seq2等。这些技术通常包含以下关键步骤:
1. 单细胞分离
2. mRNA捕获与反转录
3. cDNA扩增
4. 文库构建与测序
5. 生物信息学分析
不同技术平台在灵敏度、基因覆盖度、细胞通量和成本效益等方面各有优势,研究者应根据具体研究问题选择合适的技术方案。
第二部分:数据预处理与质量控制
高质量的数据预处理是可靠分析结果的基础。这一阶段的主要目标是过滤低质量细胞和噪声,为后续分析提供纯净的数据集。
数据加载与初步检查
import scanpy as sc
import matplotlib.pyplot as plt
import numpy as np
import pandas as pd
import seaborn as sns
import os
# 设置图形参数
sc.settings.verbosity = 3 # 输出信息详细程度
sc.settings.set_figure_params(dpi=100, figsize=(8, 6))
plt.rcParams['font.sans-serif'] = ['SimHei'] # 中文字体支持
plt.rcParams['axes.unicode_minus'] = False
# 加载数据(以PBMC数据集为例)
adata = sc.datasets.pbmc68k_reduced()
print(f"原始数据维度: {adata.shape}") # (细胞数, 基因数)
# 查看数据结构
print("数据结构:")
print(f"X矩阵: {type(adata.X)}, 形状: {adata.X.shape}")
print(f"obs表格: {adata.obs.shape}, 包含细胞注释信息")
print(f"var表格: {adata.var.shape}, 包含基因注释信息")质量控制指标计算与过滤
常用的质量控制指标包括:
1. 每个细胞检测到的基因数(越高越好,但极端值可能代表双胞或多胞)
2. 每个细胞的UMI计数(反映测序深度)
3. 线粒体基因比例(通常高比例表示细胞应激或死亡)
# 计算质量控制指标
sc.pp.calculate_qc_metrics(adata, inplace=True, percent_top=[50, 100, 200, 500])
# 绘制质量控制指标分布
fig, axs = plt.subplots(2, 2, figsize=(15, 10))
sns.histplot(adata.obs['n_genes_by_counts'], kde=True, ax=axs[0, 0])
axs[0, 0].set_title('每个细胞的基因数分布')
axs[0, 0].set_xlabel('基因数')
axs[0, 0].set_ylabel('细胞数量')
sns.histplot(adata.obs['total_counts'], kde=True, ax=axs[0, 1])
axs[0, 1].set_title('每个细胞的UMI计数分布')
axs[0, 1].set_xlabel('UMI计数')
axs[0, 1].set_ylabel('细胞数量')
# 如果有线粒体基因标记
if 'percent_mito' in adata.obs.columns:
sns.histplot(adata.obs['percent_mito'], kde=True, ax=axs[1, 0])
axs[1, 0].set_title('线粒体基因比例分布')
axs[1, 0].set_xlabel('线粒体基因比例 (%)')
axs[1, 0].set_ylabel('细胞数量')
# 散点图:基因数 vs UMI计数
sns.scatterplot(data=adata.obs, x='total_counts', y='n_genes_by_counts', ax=axs[1, 1])
axs[1, 1].set_title('基因数 vs UMI计数')
axs[1, 1].set_xlabel('UMI计数')
axs[1, 1].set_ylabel('基因数')
plt.tight_layout()
plt.savefig('quality_control_metrics.png')
plt.close()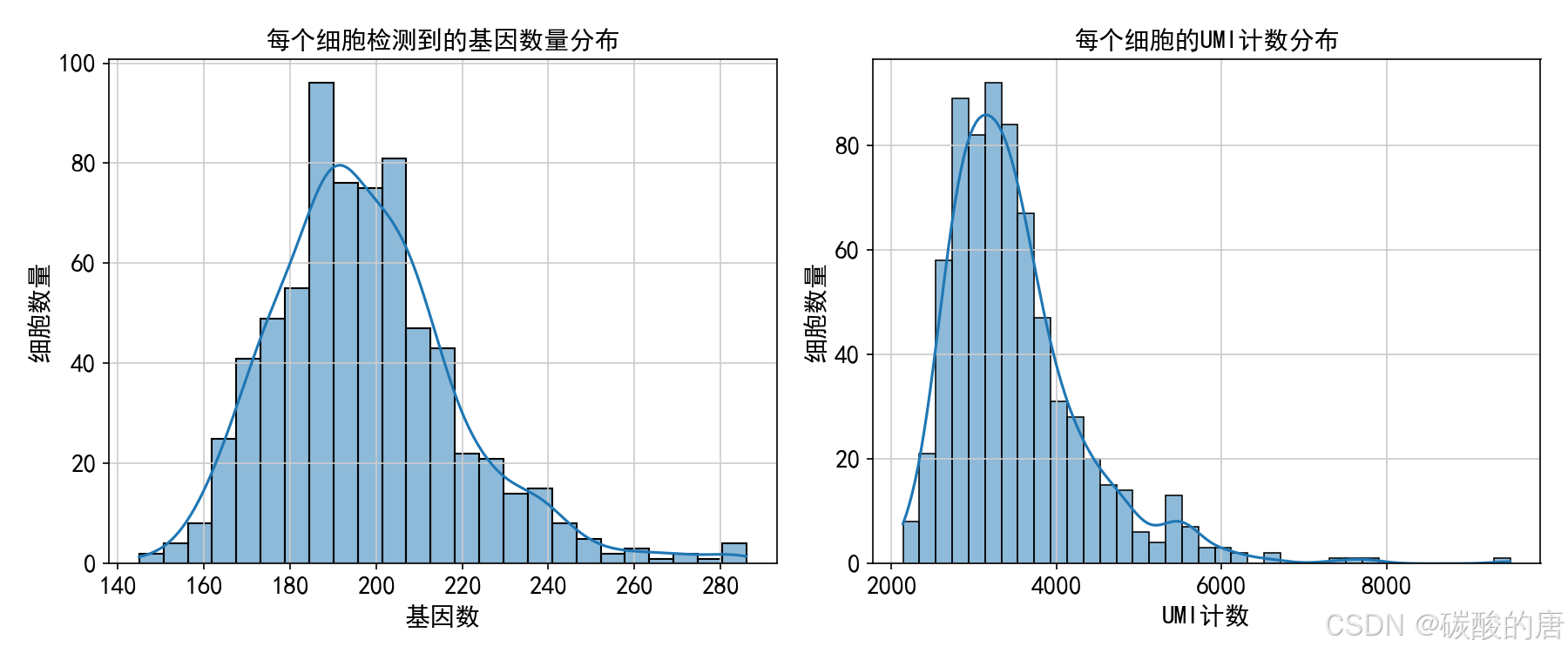
上图展示了细胞质量的关键指标:左侧为每个细胞检测到的基因数,反映测序覆盖度;右侧为每个细胞的UMI计数,反映测序深度。这些分布帮助我们设定合理的过滤阈值。
数据过滤
基于质量控制指标,我们可以过滤低质量细胞和低表达基因:
# 过滤低质量细胞
sc.pp.filter_cells(adata, min_genes=200)
print(f"过滤后的细胞数: {adata.shape[0]}")
# 过滤低表达基因
sc.pp.filter_genes(adata, min_cells=3)
print(f"过滤后的基因数: {adata.shape[1]}")
# 过滤线粒体基因比例过高的细胞
if 'percent_mito' in adata.obs.columns:
adata = adata[adata.obs.percent_mito < 20, :]
print(f"过滤线粒体后的细胞数: {adata.shape[0]}")
# 过滤潜在的双胞
adata = adata[adata.obs.n_genes_by_counts < 2500, :]
print(f"过滤潜在双胞后的细胞数: {adata.shape[0]}")数据标准化
标准化旨在消除技术变异,如测序深度差异,使细胞间比较更准确:
# 计数数据标准化(每个细胞的总计数标准化为10,000)
sc.pp.normalize_total(adata, target_sum=1e4)
# 对标准化后的数据进行对数转换
sc.pp.log1p(adata)
print("数据标准化完成")
# 可视化标准化前后的分布变化
genes_to_plot = ['CD3D', 'CD79A'] # 选择代表性基因
valid_genes = [gene for gene in genes_to_plot if gene in adata.var_names]
if valid_genes:
fig, axs = plt.subplots(1, 2, figsize=(12, 5))
for i, gene in enumerate(valid_genes):
# 提取原始表达值
if scipy.sparse.issparse(adata.raw.X):
raw_expr = adata.raw.X[:, adata.raw.var_names.get_loc(gene)].toarray().flatten()
else:
raw_expr = adata.raw.X[:, adata.raw.var_names.get_loc(gene)]
# 提取标准化后表达值
if scipy.sparse.issparse(adata.X):
norm_expr = adata.X[:, adata.var_names.get_loc(gene)].toarray().flatten()
else:
norm_expr = adata.X[:, adata.var_names.get_loc(gene)]
# 绘制原始表达值分布
sns.histplot(raw_expr[raw_expr > 0], ax=axs[i], kde=True, color='blue', label='原始数据')
# 绘制标准化后表达值分布
sns.histplot(norm_expr[norm_expr > 0], ax=axs[i], kde=True, color='red', label='标准化后')
axs[i].set_title(f'{gene}表达分布')
axs[i].set_xlabel('表达值')
axs[i].set_ylabel('细胞数量')
axs[i].legend()
plt.tight_layout()
plt.savefig('normalization_comparison.png')
plt.close()高变异基因选择
在单细胞RNA测序数据中,大多数基因表达噪声较大或无差异表达。选择高变异基因可以降低计算复杂度并提高信噪比:
# 识别高变异基因
sc.pp.highly_variable_genes(adata, n_top_genes=2000, flavor='seurat')
print(f"识别出的高变异基因数量: {sum(adata.var.highly_variable)}")
# 可视化高变异基因
plt.figure(figsize=(10, 8))
sc.pl.highly_variable_genes(adata, show=False)
plt.tight_layout()
plt.savefig('highly_variable_genes.png')
plt.close()
# 仅保留高变异基因进行后续分析
adata_hvg = adata[:, adata.var.highly_variable].copy()
print(f"仅包含高变异基因的数据形状: {adata_hvg.shape}")
# 保留原始数据作为参考
adata.raw = adata第三部分:降维分析
高维单细胞转录组数据需要降维以便于可视化和后续分析。常用的降维方法包括PCA(主成分分析)、t-SNE和UMAP。
主成分分析(PCA)
PCA是一种线性降维技术,它保留数据中最大的变异方向:
# 对数据进行缩放
sc.pp.scale(adata_hvg, max_value=10)
# 执行PCA
sc.tl.pca(adata_hvg, n_comps=50)
print(f"PCA计算完成,维度: {adata_hvg.obsm['X_pca'].shape}")
# 可视化PCA结果
fig, axs = plt.subplots(1, 3, figsize=(18, 6))
# PCA散点图
sc.pl.pca(adata_hvg, color='bulk_labels', ax=axs[0], show=False)
axs[0].set_title('PCA降维 (PC1 vs PC2)')
# PCA解释方差比例
sc.pl.pca_variance_ratio(adata_hvg, n_pcs=30, ax=axs[1], show=False)
axs[1].set_title('PCA解释方差比例')
# PCA载荷图(显示每个基因对主成分的贡献)
sc.pl.pca_loadings(adata_hvg, components=[0, 1], ax=axs[2], show=False)
axs[2].set_title('PCA基因载荷 (PC1 & PC2)')
plt.tight_layout()
plt.savefig('pca_analysis.png')
plt.close()构建细胞邻居图
在进行非线性降维之前,首先计算细胞之间的相似性关系,构建邻居图:
# 构建细胞邻居图
sc.pp.neighbors(adata_hvg, n_neighbors=15, n_pcs=30)
print("细胞邻居图构建完成")UMAP和t-SNE降维
UMAP和t-SNE是单细胞分析中常用的非线性降维算法,可以更好地保留局部结构:```python
# 计算UMAP嵌入
sc.tl.umap(adata_hvg)
print("UMAP计算完成")
# 计算t-SNE嵌入
sc.tl.tsne(adata_hvg, n_pcs=30)
print("t-SNE计算完成")
# 可视化并比较不同降维方法
fig, axs = plt.subplots(1, 3, figsize=(21, 7))
# PCA可视化
sc.pl.pca(adata_hvg, color='bulk_labels', ax=axs[0], show=False)
axs[0].set_title('PCA降维')
# UMAP可视化
sc.pl.umap(adata_hvg, color='bulk_labels', ax=axs[1], show=False)
axs[1].set_title('UMAP降维')
# t-SNE可视化
sc.pl.tsne(adata_hvg, color='bulk_labels', ax=axs[2], show=False)
axs[2].set_title('t-SNE降维')
plt.tight_layout()
plt.savefig('dimensionality_reduction_comparison.png')
plt.close()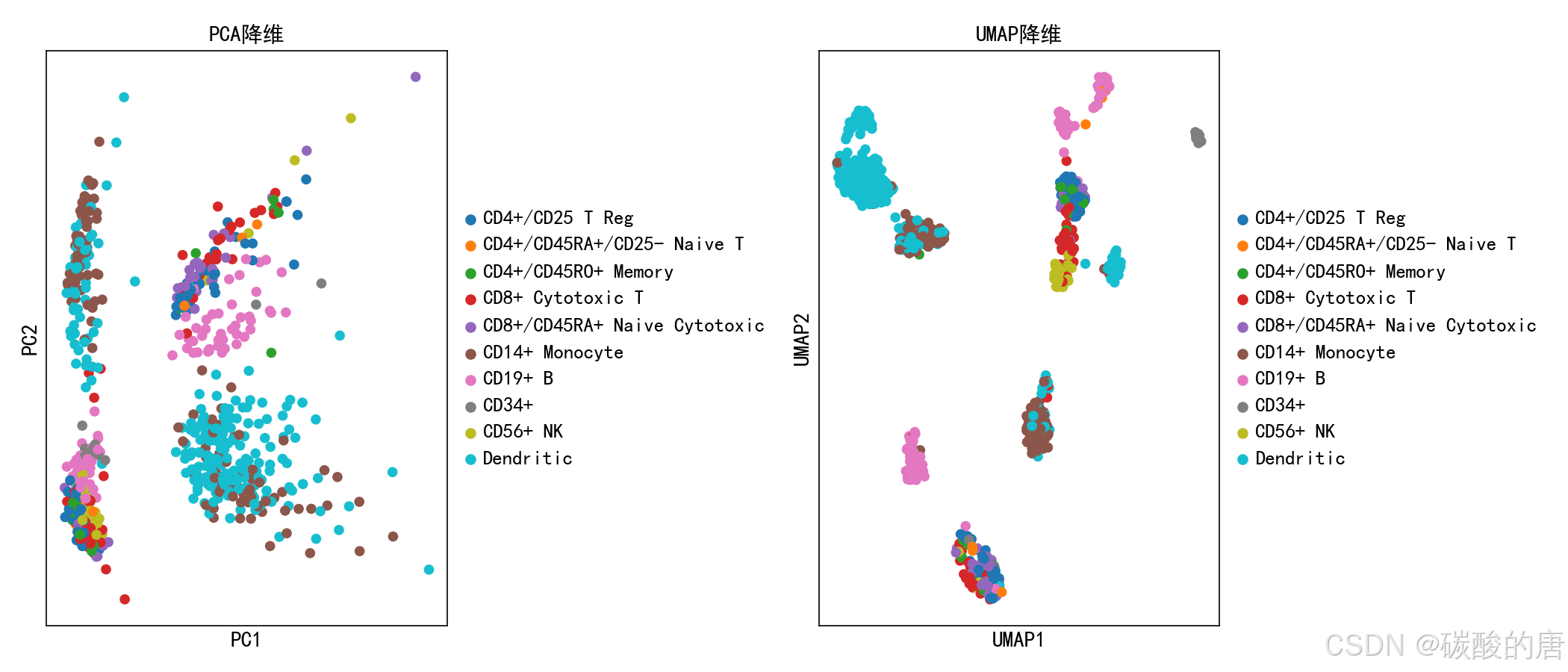
上图展示了PCA(左)和UMAP(右)两种降维方法在PBMC数据上的表现。UMAP能够更好地分离不同细胞类型,并保持局部结构。每种颜色代表一个已知的细胞类型,可以看到UMAP在区分免疫细胞亚型方面表现更佳。
 最低0.47元/天 解锁文章
最低0.47元/天 解锁文章



















 2436
2436

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











