




参考优快云博主“生信小白要知道”原文,以及Seuart官方教程。笔者鸣谢。
00 安装包与数据导入
数据集来自Seuart官方教程pbmc3k_filtered_gene_bc_matrices.tar.gz。
该步骤主要是创建初始化的Seurat对象,作为单细胞测序数据集以及后续分析结果的容器。
安装及加载所需要的包
# 安装Seurat包
BiocManager::install("Seurat")
# 加载所需的包
library(dplyr)
library(Seurat)
library(patchwork)
数据导入与创建对象
数据读取
pbmc.data <- Read10X(data.dir = "D:/生信/PBMC3Kdata") # 读取10x Genomics数据,解压后的原文件为matrix.mtx、features.tsv、barcodes.tsv,都在PBMC3Kdata文件夹下
pbmc.data 是一个稀疏矩阵(dgCMatrix),形状为 基因数(行数) × 细胞数(列数), 矩阵中的值表示的是在每个细胞(列)中检测到的每个特征(即基因,行)的分子数。
质量控制及对象初始化
创建一个Seuart的对象(pbmc) ,用于储存单细胞数据集的数据及后续的分析结果。
pbmc <- CreateSeuratObject(
counts = pbmc.data, # 原始计数矩阵
project = "pbmc", # 项目名称
min.cells = 3, # 仅保留在至少3个细胞中表达的基因
min.features = 200 # 仅保留检测到至少200个基因的细胞
)
min.cell用于过滤低检测率的基因。极少数细胞中表达的基因可能是技术噪声或测序错误,过滤后可减少数据维度并提高分析稳定性。min.features用于过滤质量不佳的细胞,表达基因过少的细胞可能是空滴(不含细胞的液滴)或 低质量细胞(RNA 降解严重),保留高质量细胞可避免干扰后续分析。
查看Seuart的对象(pbmc)
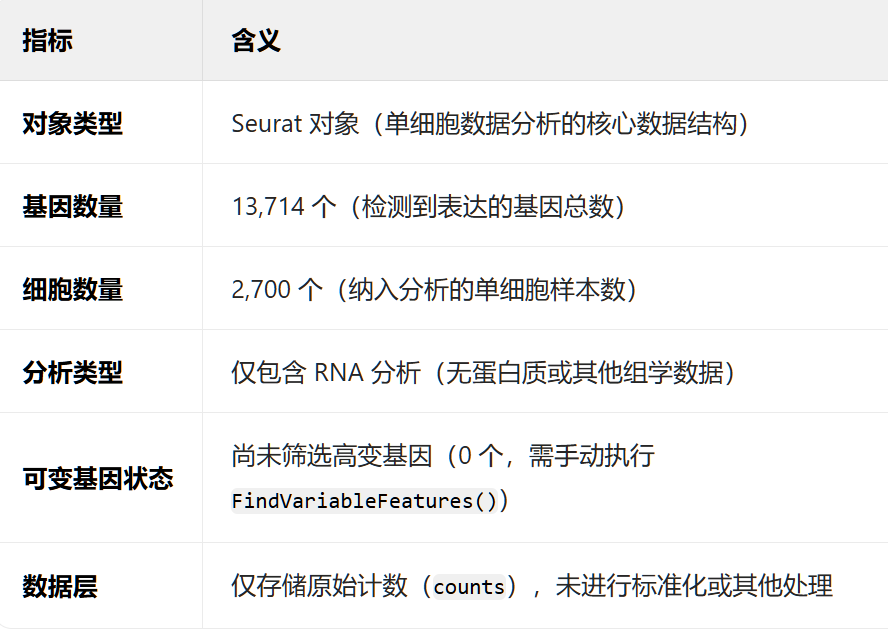
> pbmc # 查看对象摘要
An object of class Seurat
13714 features across 2700 samples within 1 assay
Active assay: RNA (13714 features, 0 variable features)
1 layer present: counts
! 关键信息总结
- 过滤后保留 13714 个基因 和 2700 个细胞。
- 当前活跃分析是 RNA,未进行变量特征筛选(0 variable features)。
01 标准预处理工作流程(质控与过滤)
该步骤主要是进行质控及选择细胞进一步分析(常用质控指标nFeature_RNA、nCount_RNA、percent.mt)
nFeature_RNA:每个细胞中检测到的基因数量。低质量的细胞或空液滴通常含有很少的基因;细胞双胞体或多胞体可能表现出异常高的基因计数。nCount_RNA:细胞内检测到的分子总数。即检测到的基因的总数量(一个基因可能会因为有多个转录本而被检测到多次),因此分子总数与基因数量密切相关。percent.mt: 对应线粒体基因组的序列的百分比。低质量/濒死的细胞通常表现出广泛的线粒体污染(细胞膜通透性增加,线粒体 DNA 释放到细胞质中被检测到)。
# 使用PercentageFeatureSet函数计算线粒体QC指标
pbmc[['percent.mt']]<-PercentageFeatureSet(pbmc,pattern = "^MT-")
设置阈值
可利用violin.plot和FeatureScatter对需质控的指标进行统计,作为后续设置阈值的参考。
violin.plot:查看质控指标数据分布特点。
小提琴形状
- 中间的箱线图:显示中位数、四分位数范围。
- 两侧的密度曲线:展示检测量的概率分布(越宽表示该区间的细胞越多)
- 图上的散点: 一个散点代表一个细胞 ,代表单个细胞的测量值. 展示了数据在细胞群组中的离散程度,补充了密度曲线无法直接反映的细节
# 使用violin plot可视化 QC指标,并使用这些指标过滤细胞
VlnPlot(pbmc,features = c("nFeature_RNA","nCount_RNA","percent.mt"),ncol = 3)

图中单个点代表一个细胞。可见单个细胞内检测到的,绝大部分基因数低于2000且大于200、绝大部分RNA数处于2000~7000、绝大染色体基因比例低于5%。
FeatureScatter:查看任意两特征之间的相关性。
plot1<-FeatureScatter(pbmc,feature1 = "nCount_RNA",feature2 = "percent.mt")
plot2<-FeatureScatter(pbmc,feature1 = "nCount_RNA",feature2 = "nFeature_RNA")
plot1+plot2

由图可见,单个细胞内检测到的RNA数目与染色体基因比例没有关系(相关性-0.13);单个细胞内检测到的RNA数目(测序深度)与基因数呈强正相关(0.95),即测序深度越深,检测到的基因数量越多。
开始过滤
可依据小提琴图和特征散点图结果选择过滤阈值。本文根据官方教程数据,过滤阈值如下
# 过滤基因数大于2500或少于200的细胞,过滤线粒体基因表达占总表达量>5%的细胞
pbmc<-subset(pbmc,subset=nFeature_RNA>200 & nFeature_RNA<2500 & percent.mt<5)
过滤完成
> # 查看过滤后的seurat数据
> pbmc
An object of class Seurat
13714 features across 2638 samples within 1 assay
Active assay: RNA (13714 features, 0 variable features)
1 layer present: counts
可见细胞数量降低到了2638个,基因数目不变。
02 数据标准化与特征选择
该步骤主要是对过滤后的数据进行标准化,主要目的是为了消除基因间表达水平的尺度差异,使不同基因在后续分析中具有可比性。
数据标准化
有LogNormalize和SCTransform两种方法,本文按照官方教程采用LogNormalize方法
- 计算方法:
Value:该基因在该细胞中的表达量
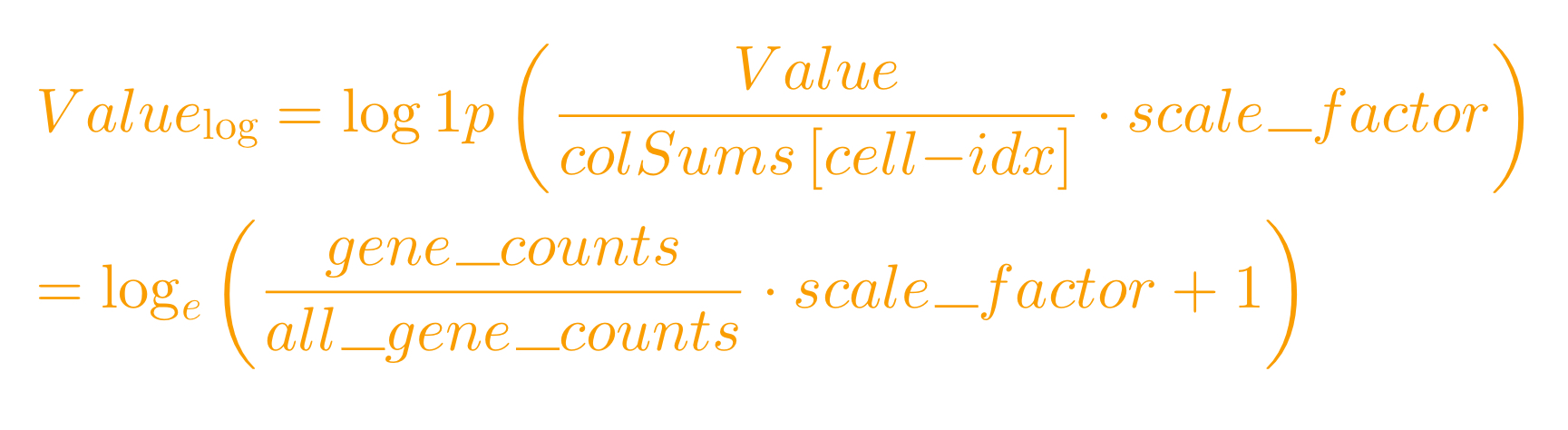
第一步:
计算每个细胞的总表达量:对于每个细胞,将该细胞中所有基因的表达计数相加,得到该细胞的总表达量。
第二步:
计算缩放后的表达量:将每个细胞中每个基因的表达计数除以该细胞的总表达量,然后乘以一个缩放因子(由 scale.factor 参数指定),得到缩放后的表达量。
第三步:
取对数变换:对缩放后的表达量取自然对数(加 1 以避免对 0 取对数),得到最终的标准化表达量。对数变换可以使数据更加符合正态分布,同时压缩数据的动态范围,减少高表达基因对分析的影响。
- 执行代码
# 数据规范化
pbmc<-NormalizeData(pbmc,normalization.method = "LogNormalize",scale.factor = 10000)
scale.factor = 10000
scale.factor 参数用于指定缩放因子。在 “LogNormalize” 方法中,将每个细胞中每个基因的表达计数除以该细胞的总表达量后,会乘以这个缩放因子。这里设置为 10000,意味着将每个细胞的基因表达量缩放为每 10000 个测序读段的表达量,这样可以使不同细胞之间的基因表达量具有统一的尺度。
特征选择(鉴定高可变基因)
使用高变基因进行主成分分析,是因为高变基因在细胞间的表达差异较大,能够更好地反映细胞的异质性,从而提高主成分分析的效果。
# 识别高度可变的特征(特征选择)
pbmc<-FindVariableFeatures(pbmc,selection.method = "vst",nfeatures = 2000)
默认返回数据集前2000个基因用于下游分析,以节省内存
> # 确定高表达的前十个基因
> top10<-head(VariableFeatures(pbmc),10)
> plot1<-VariableFeaturePlot(pbmc)
> plot2<-LabelPoints(plot = plot1,points = top10,repel = TRUE)
When using repel, set xnudge and ynudge to 0 for optimal results
> plot1+plot2

如图,红色部分代表细胞间高变基因,横轴为其在不同细胞间的平均表达量,纵轴为其在不同细胞间表达水平的标准差。图2中标注出了top10的高变基因名称。
03 数据缩放与降维
数据缩放(数据中心化)
在计算完高可变基因后,需要对数据集进行一个线性转换(缩放)。
这个步骤是降维(如PCA)数据处理之前的一个标准的预处理过程,由ScaleData()这个函数执行。
ScaleData()
- 调整每个基因的表达量,使得细胞间的平均表达量为0
- 缩放每个基因的表达量,使得细胞间的方差为1
- 目的:这一步骤赋予每个基因在下游分析中同样的权重,不至于使高表达基因占据主导地位
- 结果存储在pbmc[[“RNA”]]$scale.data中
# 以Seurat对象的行名(所有基因)抓取出来
all.genes <- rownames(pbmc)
# 以基因为单位对矩阵数据进行缩放
pbmc <- ScaleData(pbmc, features = all.genes)
概括起来讲,scale的作用是使数据具有一个统一的尺度,这并不会影响每个点的相对位置,只是使他们的表达量尺度统一起来。是PCA分析的必要步骤。
数据降维
PCA线性降维
PCA 本质上是一种无监督的线性降维技术,其核心目标是把高维数据转换为低维表示(符合单细胞数据高维稀疏的特点),同时尽可能多地保留原始数据中的信息。具体做法是通过找到数据的协方差矩阵的特征向量和特征值,其中特征向量就是主成分的方向,特征值表示对应主成分所解释的方差大小。
在进行 PCA 时,它会构建一系列相互正交(即彼此独立)的主成分。第一个主成分会沿着数据方差最大的方向,它能解释原始数据中最多的变异信息;第二个主成分则与第一个主成分正交,并且沿着剩余方差最大的方向,解释剩余变异中最多的部分,以此类推。所以,不同的主成分代表了数据中不同方向上的变异模式(即细胞类型、状态、代谢通路等区别)。
# 线性降维
pbmc<-RunPCA(pbmc,features = VariableFeatures(object=pbmc))
PC_ 1
Positive: CST3, TYROBP, LST1, AIF1, FTL, FTH1, LYZ, FCN1, S100A9, TYMP
FCER1G, CFD, LGALS1, S100A8, CTSS, LGALS2, SERPINA1, IFITM3, SPI1, CFP
PSAP, IFI30, SAT1, COTL1, S100A11, NPC2, GRN, LGALS3, GSTP1, PYCARD
Negative: MALAT1, LTB, IL32, IL7R, CD2, B2M, ACAP1, CD27, STK17A, CTSW
CD247, GIMAP5, AQP3, CCL5, SELL, TRAF3IP3, GZMA, MAL, CST7, ITM2A
MYC, GIMAP7, HOPX, BEX2, LDLRAP1, GZMK, ETS1, ZAP70, TNFAIP8, RIC3
PC_ 2
Positive: CD79A, MS4A1, TCL1A, HLA-DQA1, HLA-DQB1, HLA-DRA, LINC00926, CD79B, HLA-DRB1, CD74
HLA-DMA, HLA-DPB1, HLA-DQA2, CD37, HLA-DRB5, HLA-DMB, HLA-DPA1, FCRLA, HVCN1, LTB
BLNK, P2RX5, IGLL5, IRF8, SWAP70, ARHGAP24, FCGR2B, SMIM14, PPP1R14A, C16orf74
Negative: NKG7, PRF1, CST7, GZMB, GZMA, FGFBP2, CTSW, GNLY, B2M, SPON2
CCL4, GZMH, FCGR3A, CCL5, CD247, XCL2, CLIC3, AKR1C3, SRGN, HOPX
TTC38, APMAP, CTSC, S100A4, IGFBP7, ANXA1, ID2, IL32, XCL1, RHOC
PC_ 3
Positive: HLA-DQA1, CD79A, CD79B, HLA-DQB1, HLA-DPB1, HLA-DPA1, CD74, MS4A1, HLA-DRB1, HLA-DRA
HLA-DRB5, HLA-DQA2, TCL1A, LINC00926, HLA-DMB, HLA-DMA, CD37, HVCN1, FCRLA, IRF8
PLAC8, BLNK, MALAT1, SMIM14, PLD4, P2RX5, IGLL5, LAT2, SWAP70, FCGR2B
Negative: PPBP, PF4, SDPR, SPARC, GNG11, NRGN, GP9, RGS18, TUBB1, CLU
HIST1H2AC, AP001189.4, ITGA2B, CD9, TMEM40, PTCRA, CA2, ACRBP, MMD, TREML1
NGFRAP1, F13A1, SEPT5, RUFY1, TSC22D1, MPP1, CMTM5, RP11-367G6.3, MYL9, GP1BA
PC_ 4
Positive: HLA-DQA1, CD79B, CD79A, MS4A1, HLA-DQB1, CD74, HIST1H2AC, HLA-DPB1, PF4, SDPR
TCL1A, HLA-DRB1, HLA-DPA1, HLA-DQA2, PPBP, HLA-DRA, LINC00926, GNG11, SPARC, HLA-DRB5
GP9, AP001189.4, CA2, PTCRA, CD9, NRGN, RGS18, CLU, TUBB1, GZMB
Negative: VIM, IL7R, S100A6, IL32, S100A8, S100A4, GIMAP7, S100A10, S100A9, MAL
AQP3, CD2, CD14, FYB, LGALS2, GIMAP4, ANXA1, CD27, FCN1, RBP7
LYZ, S100A11, GIMAP5, MS4A6A, S100A12, FOLR3, TRABD2A, AIF1, IL8, IFI6
PC_ 5
Positive: GZMB, NKG7, S100A8, FGFBP2, GNLY, CCL4, CST7, PRF1, GZMA, SPON2
GZMH, S100A9, LGALS2, CCL3, CTSW, XCL2, CD14, CLIC3, S100A12, RBP7
CCL5, MS4A6A, GSTP1, FOLR3, IGFBP7, TYROBP, TTC38, AKR1C3, XCL1, HOPX
Negative: LTB, IL7R, CKB, VIM, MS4A7, AQP3, CYTIP, RP11-290F20.3, SIGLEC10, HMOX1
LILRB2, PTGES3, MAL, CD27, HN1, CD2, GDI2, CORO1B, ANXA5, TUBA1B
FAM110A, ATP1A1, TRADD, PPA1, CCDC109B, ABRACL, CTD-2006K23.1, WARS, VMO1, FYB
> print(pbmc[["pca"]],dims = 1:5,nfeatures = 5)
PC_ 1
Positive: CST3, TYROBP, LST1, AIF1, FTL
Negative: MALAT1, LTB, IL32, IL7R, CD2
PC_ 2
Positive: CD79A, MS4A1, TCL1A, HLA-DQA1, HLA-DQB1
Negative: NKG7, PRF1, CST7, GZMB, GZMA
PC_ 3
Positive: HLA-DQA1, CD79A, CD79B, HLA-DQB1, HLA-DPB1
Negative: PPBP, PF4, SDPR, SPARC, GNG11
PC_ 4
Positive: HLA-DQA1, CD79B, CD79A, MS4A1, HLA-DQB1
Negative: VIM, IL7R, S100A6, IL32, S100A8
PC_ 5
Positive: GZMB, NKG7, S100A8, FGFBP2, GNLY
Negative: LTB, IL7R, CKB, VIM, MS4A7
降维结果分析
可对降维结果进行可视化分析,根据Seuart官方教程有VizDimReduce()、DimPlot()和DimHeatmap()
VizDimReduce()
此函数用于可视化主成分分析(PCA)中指定主成分的基因载荷。基因载荷表示每个基因对主成分的贡献程度,通过可视化基因载荷,可以了解哪些基因对特定主成分的影响较大。
VizDimLoadings(pbmc,dims = 1:2,reduction = "pca")

图1和图2分别展示了第一和第二个主成分中,不同基因对其的影响
DimPlot()
该函数用于绘制单细胞数据在指定降维空间中的散点图,通过散点图可以直观地看到细胞在降维空间中的分布情况,有助于发现细胞亚群的聚类结构。在散点图中,每个点代表一个单细胞,不同颜色或形状的点表示不同的细胞类型或状态。通过观察散点图,我们可以发现细胞之间的聚类模式、相似性和差异性,进而对细胞类型或状态进行分类和注释。此外,DimPlot函数还可以根据其他降维方法(如t-SNE和UMAP)进行可视化,以便于我们比较不同降维方法的效果。
DimPlot(pbmc,reduction = "pca")

DimHeatmap()
这个函数用于绘制指定主成分的基因表达热图,展示与特定主成分相关的基因在部分细胞中的表达情况。热图可以帮助我们直观地观察基因表达的模式和细胞之间的相似性。在热图中,每一列代表一个细胞,每一行代表一个基因或基因集,颜色的深浅表示该基因或基因集在该细胞中的表达水平。通过观察热图,我们可以发现不同基因或基因集之间的相关性、表达模式和差异性,进而对细胞类型或状态进行分类和注释。
黄 - 红(高值区强化暖色):
- 紫色,黑色 → 低表达
- 黄色 → 高表达
DimHeatmap(pbmc,dims = 1,cells = 500,balanced = TRUE)
DimHeatmap(pbmc,dims = 1:15,cells=500,balanced = TRUE)

第一个主成分的基因表达热图

前15个主成分的基因表达热图
确定后续分析所需维度
对单细胞 RNA 测序数据进行主成分分析(PCA)后的主成分显著性评估,根据Seuart官方可使用肘形图(ElbowPlot)或JackStraw图
肘形图(ElbowPlot)更为直观,但其实际也来自于JackStraw图
JackStrawPlot()
- JackStraw() 函数:Seurat 包中用于执行 JackStraw 分析的函数。JackStraw 分析是一种用于评估主成分分析中每个主成分显著性的方法,通过随机重采样的方式来模拟随机背景分布,然后将真实的主成分与随机背景进行比较,从而判断每个主成分是否包含真实的生物学信号。
- pbmc:是一个 Seurat 对象,包含了单细胞 RNA 测序数据以及之前进行主成分分析(PCA)后的结果。
- num.replicate = 100:指定随机重采样的次数,这里设置为 100 次。在每次重采样过程中,会随机打乱基因表达数据,然后重新计算主成分,通过多次重采样可以得到一个随机背景下主成分的分布情况。
pbmc<-JackStraw(pbmc,num.replicate = 100)
pbmc<-ScoreJackStraw(pbmc,dims = 1:20)
JackStrawPlot(pbmc,dims = 1:15)

ElbowPlot(pbmc)
- ScoreJackStraw() 函数:Seurat 包中用于对 JackStraw 分析结果进行评分的函数。该函数会根据之前 JackStraw 重采样得到的随机背景分布,计算每个主成分的显著性得分(通常是 p 值),以判断该主成分是否显著。
- pbmc:同样是包含 JackStraw 分析结果的 Seurat 对象。
- dims = 1:20:指定要进行评分的主成分范围,这里表示对前 20 个主成分(PC1 - PC20)进行评分。通过评分可以筛选出那些具有显著生物学意义的主成分,用于后续的分析,如细胞聚类、差异表达分析等。
ElbowPlot(pbmc)

一般选择肘点附近(即本图中PC=10左右),作为数据集的PC值
来自优快云博主“生信小白要知道”的观点:
横坐标的PC是你接下来要选择分析的维度数,即多少组基因表达pattern。当选择一组的时候,即所有基因都在一个表达pattern里面(我们从生物学角度看也肯定是不可能的),我们很容易想到,所有细胞都在一个组内,肯定是存在很大差异的,所以方差值(方差=标准差(Standard Deviation)的平方)就会很大。当PC增多,即分组增多后,每个组内的差异也就慢慢变小了,方差值自然也就变小了。再往后,随着选的PC的增多,基本上区分不出来组内的差异了,这个时候方差基本就稳定了下来,此时在选取更大的PC值也就没有更大的意义了,并且还会增加计算压力。因此,个人认为关于PC选取的最合适原则是在拐点右侧3-5个维度即可(本数据拐点在7-8左右,我们选10刚刚好),宽松一点的话,可以设置在拐点右侧10以内。不过最重要的一点还是尽可能地多尝试,选取最适合自己的PC值,这一点也是官网教程所建议的。
04 细胞聚类与可视化
细胞聚类
FindNeighbors()根据肘部图选择前10个PC成分作为输入(dims = 1:10)
FindNeighbors 是 Seurat 包中用于构建最近邻图和共享最近邻图的函数。其基于主成分分析(PCA)的结果,计算每个细胞在特征空间中的最近邻细胞,构建最近邻图和共享最近邻图(KNN),为后续的细胞聚类分析做准备。
# 建立KNN图,并基于其局部领域中的共享重叠细化任意两个单元之间的边权重
pbmc<-FindNeighbors(pbmc,dims = 1:10)
FindClusters()
Seurat应用模块化优化技术对细胞进行聚类,如Louvain算法(默认)或 SLM [SLM, Blondel et al., Journal of Statistical Mechanics],以迭代方式将单元分组在一起,以优化标准模块化函数。该函数通过 resolution 参数设置下游聚类的“粒度”,该值的增加会导致集群数量增加。将此参数设置在 0.4-1.2 之间通常对于大约 3K 细胞的单细胞数据集会返回良好的结果。对于较大的数据集,最佳分辨率通常会提高。
pbmc<-FindClusters(pbmc,resolution = 0.5)
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 2638
Number of edges: 95927
Running Louvain algorithm...
0% 10 20 30 40 50 60 70 80 90 100%
[----|----|----|----|----|----|----|----|----|----|
**************************************************|
Maximum modularity in 10 random starts: 0.8728
Number of communities: 9
Elapsed time: 0 seconds
> head(Idents(pbmc),5)
AAACATACAACCAC-1 AAACATTGAGCTAC-1 AAACATTGATCAGC-1 AAACCGTGCTTCCG-1
2 3 2 1
AAACCGTGTATGCG-1
6
Levels: 0 1 2 3 4 5 6 7 8
关键参数
- pbmc:输入的 Seurat 对象,需提前完成 PCA 降维(如RunPCA())。
- resolution = 0.5:
控制聚类的精细程度(值越大,聚类越多)。
典型范围:0.4-1.2(数据量越大,值可越高)。- 算法原理
使用Louvain 算法优化模块度(Modularity),将相似细胞划分到同一簇。
模块度:衡量网络(细胞 - 细胞相似性)中社区结构强度的指标,值越接近 1 表示聚类质量越好。
非线性降维(UMAP/tSNE)
Seurat 提供了几种非线性降维技术,例如 tSNE 和 UMAP,用于可视化和探索这些数据集。这些算法的目标是学习数据的基础流形,以便将相似的单元放在低维空间中。
| 步骤 | 核心作用 | 不可或缺的原因 |
|---|---|---|
| PCA | 提取全局线性变异,降噪、降维,为后续分析提供低维 “干净” 数据 | 高维原始数据无法直接输入 UMAP/tSNE(计算量太大、噪声干扰强) |
| UMAP/tSNE | 在低维空间中还原数据的非线性流形结构,用于可视化亚群聚类和分布 | PCA 无法捕捉局部非线性关系(如亚群间的精细边界),而这正是单细胞分析的核心目标 |
上面确定的基于图形的聚类中的细胞应该在这些降维图上共定位。作为 UMAP 和 tSNE 的输入,按照官网建议使用相同的 PC(即依旧是前10个主成分,dims = 1:10)作为聚类分析的输入。
- UMAP
pbmc<-RunUMAP(pbmc,dims = 1:10)
DimPlot(pbmc,reduction = "umap")

- tSNE
pbmc <- RunTSNE(pbmc, dims = 1:10)
DimPlot(pbmc, reduction = "tsne")

05 差异基因分析与注释
寻找差异表达基因
Seurat 可以找到通过差异表达定义簇的标记物。默认情况下,与所有其他细胞簇相比,它识别单个簇(在 ident.1中指定)的正标记和负标记。 FindAllMarkers()为所自动执行此过程,但也可以测试细胞簇彼此之间或所有细胞之间的对比。
计算所有簇的markers
- Seurat教程仅设置了一个参数,就是only.pos,该参数的意思是只保留正标记(positive markers),也就是只保留相对于其他簇而言,目标簇显著高表达的基因标记。
- FindAllMarkers() 函数:这是 Seurat 包中的一个函数,用于对 Seurat 对象 pbmc 中的所有细胞聚类进行差异表达分析,找出每个聚类的标记基因。
- only.pos = TRUE:该参数指定只返回在特定聚类中高表达的基因,即正差异表达的基因,忽略那些在其他聚类中高表达的基因。
-min.pct = 0.25:是一个过滤条件,要求目标基因至少在 25% 的当前聚类细胞或至少 25% 的其他聚类细胞中表达,才会被纳入差异表达分析,这样可以减少在极少数细胞中表达的基因对分析结果的干扰。- logfc.threshold = 0.25:表示只考虑平均对数 2 倍变化值大于 0.25 的基因,即该基因在当前聚类中的表达量至少是其他聚类的 2^0.25 倍,这有助于筛选出表达差异更显著的基因。
> # 找出每个细胞簇的标记物,与所有剩余的细胞进行比较,只报告阳性细胞
> pbmc.markers<-FindAllMarkers(pbmc,only.pos = TRUE,min.pct = 0.25,logfc.threshold =0.25 )
Calculating cluster 0
|++++++++++++++++++++++++++++++++++++++++++++++++++| 100% elapsed=02s
Calculating cluster 1
|++++++++++++++++++++++++++++++++++++++++++++++++++| 100% elapsed=03s
Calculating cluster 2
|++++++++++++++++++++++++++++++++++++++++++++++++++| 100% elapsed=02s
Calculating cluster 3
|++++++++++++++++++++++++++++++++++++++++++++++++++| 100% elapsed=01s
Calculating cluster 4
|++++++++++++++++++++++++++++++++++++++++++++++++++| 100% elapsed=02s
Calculating cluster 5
|++++++++++++++++++++++++++++++++++++++++++++++++++| 100% elapsed=05s
Calculating cluster 6
|++++++++++++++++++++++++++++++++++++++++++++++++++| 100% elapsed=03s
Calculating cluster 7
|++++++++++++++++++++++++++++++++++++++++++++++++++| 100% elapsed=07s
Calculating cluster 8
|++++++++++++++++++++++++++++++++++++++++++++++++++| 100% elapsed=02s
- %>%:这是管道操作符,用于将前一个函数的输出作为后一个函数的输入,使代码更具可读性和简洁性。
- group_by(cluster):按照 cluster 列对 pbmc.markers 数据框进行分组,即将数据按照不同的聚类进行划分。
- slice_max(n = 2, order_by = avg_log2FC):对于每个分组(即每个聚类),按avg_log2FC 列的值进行降序排序,并选取前 2 行,也就是每个聚类中 avg_log2FC 最高的前 2 个基因。
> pbmc.markers %>%
+ group_by(cluster) %>%
+ slice_max(n=2,order_by = avg_log2FC)
# A tibble: 18 × 7
# Groups: cluster [9]
p_val avg_log2FC pct.1 pct.2 p_val_adj cluster gene
<dbl> <dbl> <dbl> <dbl> <dbl> <fct> <chr>
1 9.57e- 88 2.40 0.447 0.108 1.31e- 83 0 CCR7
2 1.35e- 51 2.14 0.342 0.103 1.86e- 47 0 LEF1
3 7.07e-139 7.28 0.299 0.004 9.70e-135 1 FOLR3
4 3.38e-121 6.74 0.277 0.006 4.64e-117 1 S100A12
5 2.97e- 58 2.09 0.42 0.111 4.07e- 54 2 AQP3
6 5.03e- 34 1.87 0.263 0.07 6.90e- 30 2 CD40LG
7 2.40e-272 7.38 0.564 0.009 3.29e-268 3 LINC00926
8 2.75e-237 7.14 0.488 0.007 3.76e-233 3 VPREB3
9 7.25e-165 4.41 0.577 0.055 9.95e-161 4 GZMK
10 3.27e- 88 3.74 0.419 0.061 4.48e- 84 4 GZMH
11 8.23e-168 5.88 0.37 0.005 1.13e-163 5 CKB
12 1.69e-212 5.43 0.506 0.01 2.32e-208 5 CDKN1C
13 8.10e-179 6.22 0.471 0.013 1.11e-174 6 AKR1C3
14 5.38e-112 6.07 0.29 0.007 7.38e-108 6 SH2D1B
15 1.46e-207 8.03 0.5 0.002 2.00e-203 7 SERPINF1
16 1.48e-220 7.63 0.812 0.011 2.03e-216 7 FCER1A
17 0 14.4 0.615 0 0 8 LY6G6F
18 7.32e-222 13.9 0.385 0 1.00e-217 8 RP11-879F14.2
计算特定簇的markers
计算单个簇对比其他所有簇的markers
# 寻找cluster2的所有markers
> cluster2.markers<-FindMarkers(pbmc,ident.1 = 2,min.pct = 0.25)
|++++++++++++++++++++++++++++++++++++++++++++++++++| 100% elapsed=06s
>head(cluster2.markers,n=5)
p_val avg_log2FC pct.1 pct.2 p_val_adj
IL32 2.892340e-90 1.3070772 0.947 0.465 3.966555e-86
LTB 1.060121e-86 1.3312674 0.981 0.643 1.453850e-82
CD3D 8.794641e-71 1.0597620 0.922 0.432 1.206097e-66
IL7R 3.516098e-68 1.4377848 0.750 0.326 4.821977e-64
LDHB 1.642480e-67 0.9911924 0.954 0.614 2.252497e-63
计算某个簇对比其他特定簇的markers
> # 寻找cluster5中与cluster0和cluster3n不同的所有markers
> cluster5.markers<-FindMarkers(pbmc,ident.1 = 5,ident.2=c(0,3),min.pct = 0.25)
|++++++++++++++++++++++++++++++++++++++++++++++++++| 100% elapsed=05s
> head(cluster5.markers,n=5)
p_val avg_log2FC pct.1 pct.2 p_val_adj
FCGR3A 8.246578e-205 6.794969 0.975 0.040 1.130936e-200
IFITM3 1.677613e-195 6.192558 0.975 0.049 2.300678e-191
CFD 2.401156e-193 6.015172 0.938 0.038 3.292945e-189
CD68 2.900384e-191 5.530330 0.926 0.035 3.977587e-187
RP11-290F20.3 2.513244e-186 6.297999 0.840 0.017 3.446663e-182
基因表达可视化
Vlnplot(显示跨集群的表达概率分布)和FeaturePlot()(在tSNE或PCA图上可视化特征表达)是最常用的可视化
violinplot
- 横轴(X 轴)
含义:通常代表细胞簇(Cluster),即之前通过 FindClusters() 函数得到的聚类结果。
例如,横轴可能显示 0, 1, 2, … 或 B_cell, T_cell, Macrophage 等(取决于是否已注释细胞类型)。- 纵轴(Y 轴)
含义:基因的表达量(通常是标准化后的对数表达值,如 log2(TPM + 1))。
# 展示特定基因表达水平
VlnPlot(pbmc,features = c("MS4A1","CD79A"))

可见"MS4A1","CD79A"基因在不同细胞簇中的表达量分布,用于识别哪些簇是 B 细胞(高表达这两个基因的簇,即3号簇)。
# 可以绘制行数据
VlnPlot(pbmc,features = c("NKG7","PF4"),slot = "counts",log = TRUE)
- slot = "counts"的特殊性:
原始计数数据可能存在零膨胀(许多基因在多数细胞中不表达)。
对数转换后,零值变为 log2 (0+1)=0,但高表达值被压缩,可能弱化差异。
适用于展示绝对表达量差异,但需注意数据分布偏态。
- log 参数的影响(log参数用于控制是否对基因表达量进行对数转换)
log = TRUE:压缩高表达值,突出低表达区域的差异(更常见)。
log = FALSE:直接展示原始计数,但可能因数据偏态导致图形失真。

可见"NKG7","PF4"分别在不同细胞簇中的表达水平(NKG7 高表达 → NK 细胞或细胞毒性 T 细胞,即4号簇;PF4 高表达 → 血小板或巨核细胞,即8号簇)。
FeaturePlot()
# 多个特征图
FeaturePlot(pbmc,features = c("MS4A1", "GNLY", "CD3E", "CD14", "FCER1A", "FCGR3A", "LYZ", "PPBP", "CD8A"))

展示features参数中的基因在UMAP细胞聚类中,不同表达水平细胞的具象化分布(单个点代表细胞,点的颜色深浅代表细胞中该基因的表达水平)
DoHeatmap()
#DoHeatmap()为给定的细胞和特征生成一个表达热图。在这种情况下,我们为每个集群绘制前 20 个标记(或所有标记,如果小于 20)。
pbmc.markers%>%
group_by(cluster)%>%
top_n(n=10,wt=avg_log2FC)->top10
DoHeatmap(pbmc,features = top10$gene)+NoLegend()
- 横轴:细胞(按簇分组,如簇 0-8)。
- 纵轴:基因(按簇分组,每个簇的 Top 10 基因)。

细胞簇命名
# 将细胞类型标识分配给集群
new.cluster.ids<-c("Naive CD4 T", "CD14+ Mono", "Memory CD4 T", "B", "CD8 T", "FCGR3A+ Mono",
"NK", "DC", "Platelet")
names(new.cluster.ids)<-levels(pbmc)
pbmc<-RenameIdents(pbmc,new.cluster.ids)
DimPlot(pbmc,reduction = "umap",label = TRUE,pt.size = 0.5)+NoLegend()
saveRDS(pbmc,file = "./pbmc3k_final.rds")


















 9982
9982

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言










{kind=link}