










本人是
win11,薛定谔版本是12.9。
官网:https://www.schrodinger.com/
本篇文章的示例大分子蛋白PDB ID为4KNN,小分子配体的MOL ID为MOL004004。
本文部分图源来自知乎https://zhuanlan.zhihu.com/p/416698194,推荐为原作者贡献阅读量捏。
1.Ligand docking讲解(可跳过)
在右上角的操作区的tasks搜索Ligand docking,打开面板如下:
1.1.Ligands

1.2.Settings

HTVS:高通量筛选,一般用来筛选很多个小分子SP:和HTVS算法原理差不多,但是会降低采样的彻底性。XP:开始和SP是一样的,但是采样更严格,运行时间比SP长。对于形状互补更严格,所以一般用于排除假阳性。但是需要额外的license
补充:如果你需要虚拟筛选一个很大的数据库,建议先用
SP,按照打分排序,取前10%-30%,用XP重新对接。并勾选write xp descriptor information。
还有一个virtual screening模块,相当于先用HTVS、再sp、再xp。
1.3.Output

1.4.job settings

2.Ligand docking实战
在右上角的操作区的tasks搜索Ligand docking,打开面板如下:
默认从文件中加载glide-grid1.zip并勾选display receptor 和show grid boxes。
选择配体:use ligands from files,并加载ligprep_1-out.maegz。最后直接run即可。

对接完成后会得到glide-dock_SP_1_pv.maegz文件,且左侧工作区会生成多个名字和小分子配体名字一样的条目,实际上对接后的构象,在我这是MOL004004。
我们点击操作区的table,显示如下,只用看docking score这一列。

可以看到,最好的结果是-5.559,只能说有作用力,但对接得不算特别好。
3.可视化
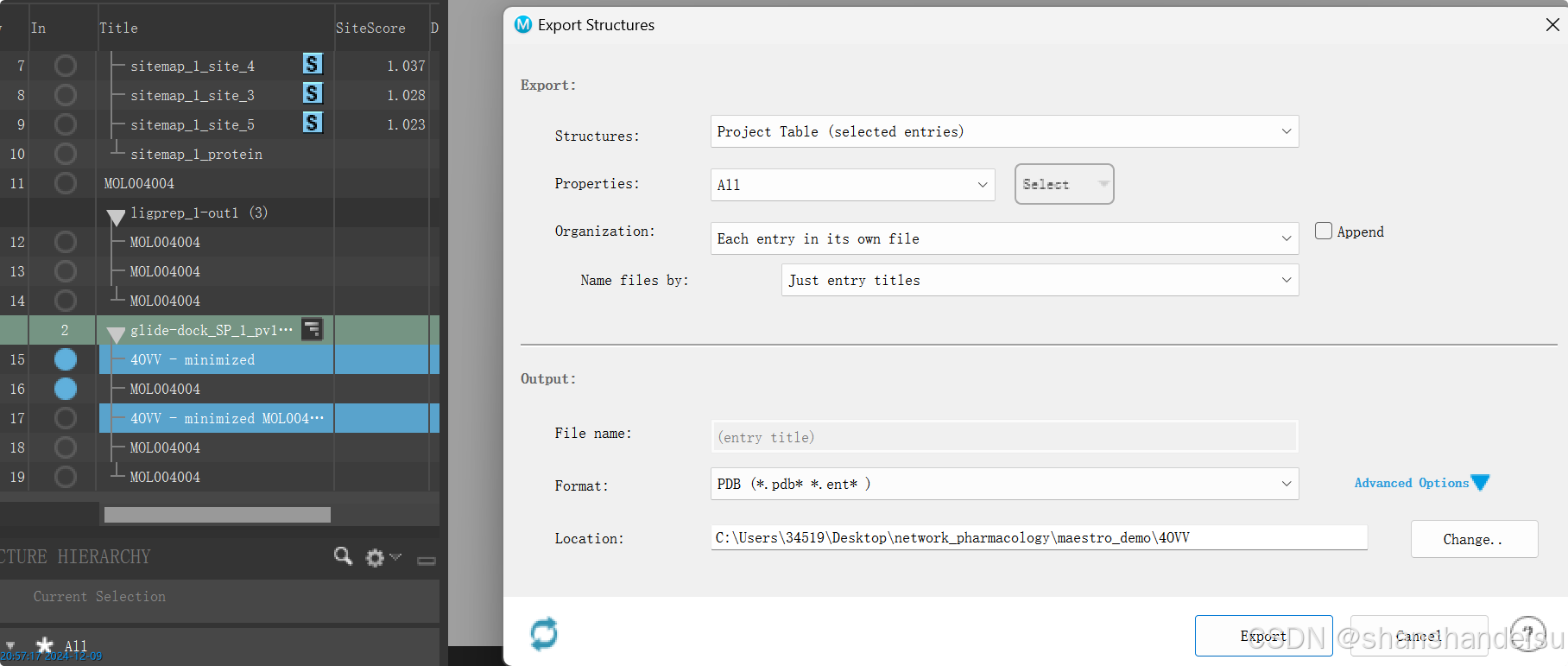
3.1.导出蛋白配体复合物
选中结合能最高的条目和minimized蛋白质条目,右键merge得到蛋白配体复合物,在这就是4OVV - minimized MOL004004。
然后多选,minimized蛋白质条目,蛋白配体复合物,这2个条目,右键export structures导出为PDB格式。
3.2.分析2D Sketcher
以下都是25.03.21日补更的内容,所以蛋白变为了6YR9,配体变为了MOL003963。
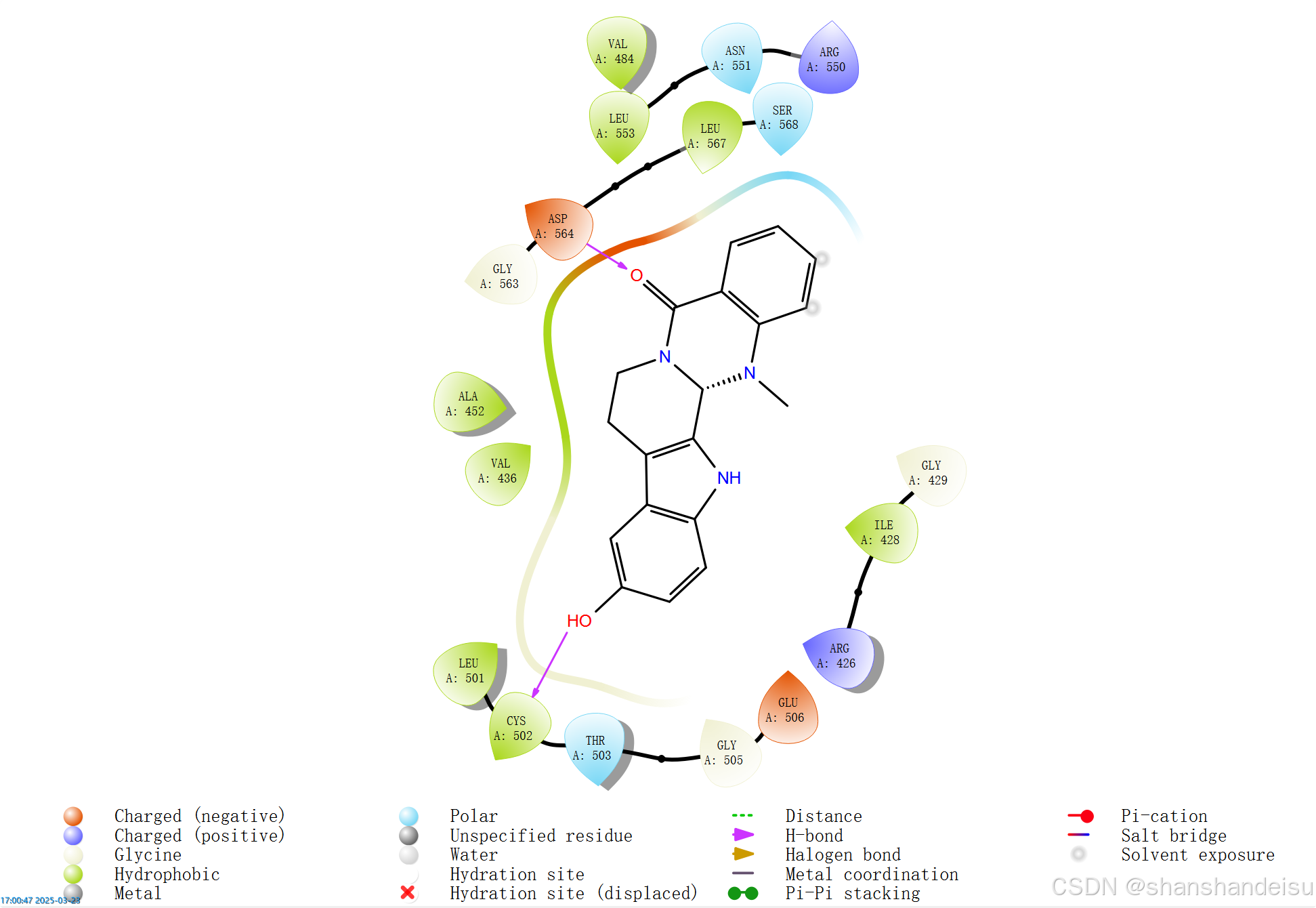
我们需要选中这个蛋白配体复合物,然后在tasks中找到2D Sketcher,点击后它会对接后的蛋白配体复合物的2D作用图。
打开View -> LID Legend,可以查看颜色和键的对应。
而且待会保存图片时才会保存这些批注。

默认:紫色的是氢键,也就是这里140位残基的两个键和85位残基的一个键。
绿色的是Π键,也就是这里81位的一个键。
注意,和我文章
2、基础使用提到的默认颜色对应的是不同的哈。
你也可以再点击下左侧栏的best 2D view,也就是⭐样式的图标

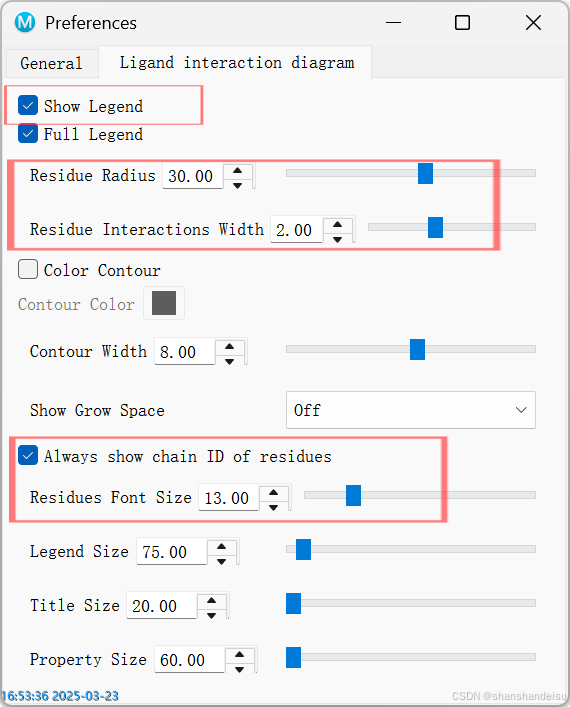
如果你觉得标签字体太小的话,可以点击edit/preference,改动如下选项:
最终如下:
选择File/Save ScreenShot,注意不要勾选Transparent Background,我一般会将width调整为1200,点击ok导出图片。我一般习惯命名为2D.png。
3.3.在pymol中处理
3.3.1.整体输出
然后将复合物导入pymol打开,进行一些通用设置。
- 打开残基序列,分离蛋白和配体,并分别命名为
pro和lig。 - 蛋白颜色设置为
cyans/palecyan。 - 配体条目选择
Hide/Valence。 - 调整标签大小
setting/label/size/18point - 调整背景颜色为白色。
- 保存工程文件
File/Save Session As一般我就命名为pymol.pse
然后在pymol命令行下输入如下命令:
select res, chain A and resi 564 + chain A and resi 502
这里的数字和链名要根据你自己刚刚2D作用图更改。
如果只有一个,譬如这里我只有一个140的话,就是
select res, chain A and resi 564
回车运行后,发现作用的残基就已经被找到了,而且被命名为res
我们将res颜色配置为yellow/paleyellow,并且将样式改为stick。
在命令行输入以下命令,隐藏多余的水分子。
hide sticks, h.
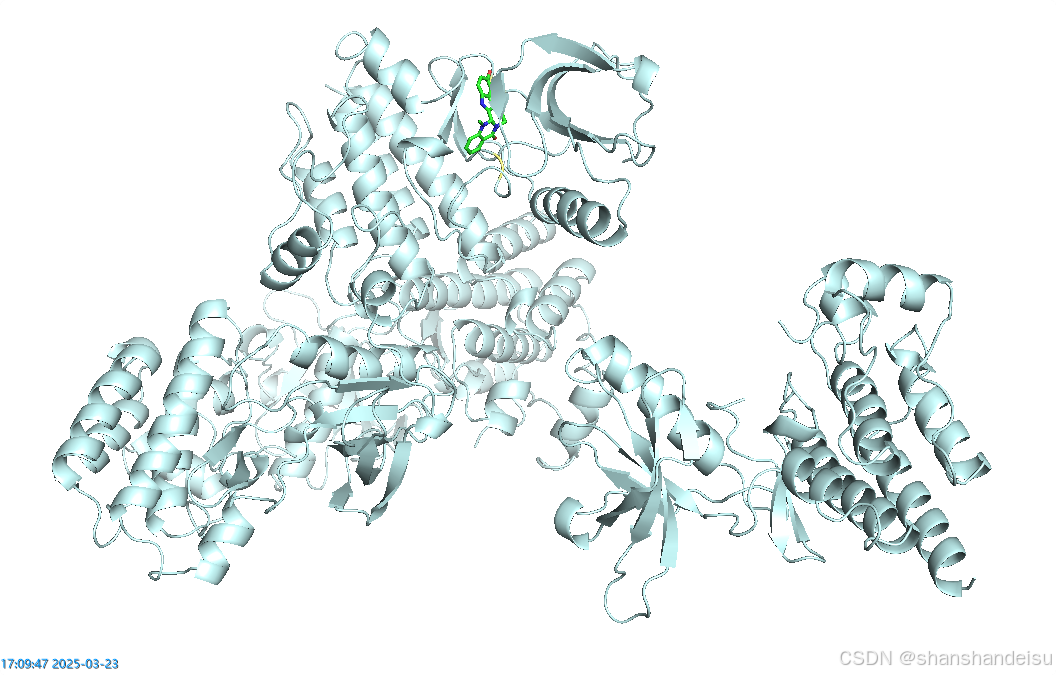
接下来我们就可以输出一张整体的图片了。
选择右上角的Draw/Ray,调整为300dpi,并且取消勾选transparent,选择draw(faset),选择保存为文件。
命名我一般习惯为big.png(注意,配体放中间偏左下会好看一点)
3.3.2.细节输出
选择settings/transparency/cartoon,调整为60%。
然后选中res条目,调整样式为stick。并且L/residues打开标签。
左上角调整为原子模式。
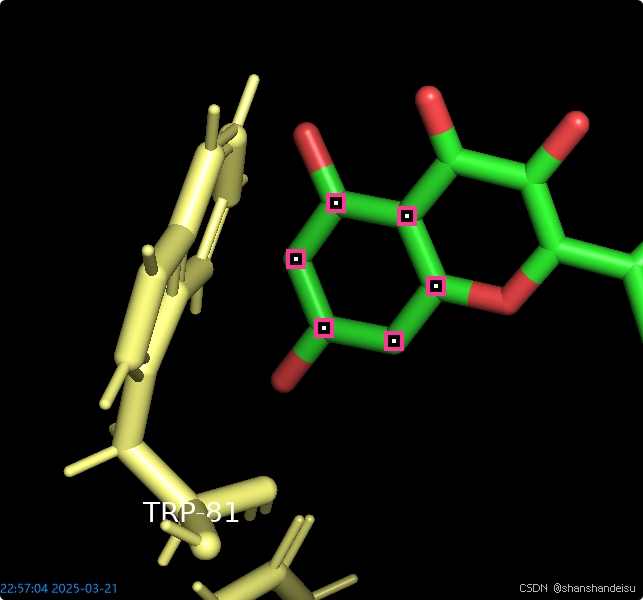
如果有作用在苯环中心的键,需要定义苯环平面
在命令行输入命令:pseudoatom p1,sele
这个命令的意思是定义一个平面并命名为p1,我们就可以看到苯环中间出现了一个标志,而且右边层级多了一个p1的条目。
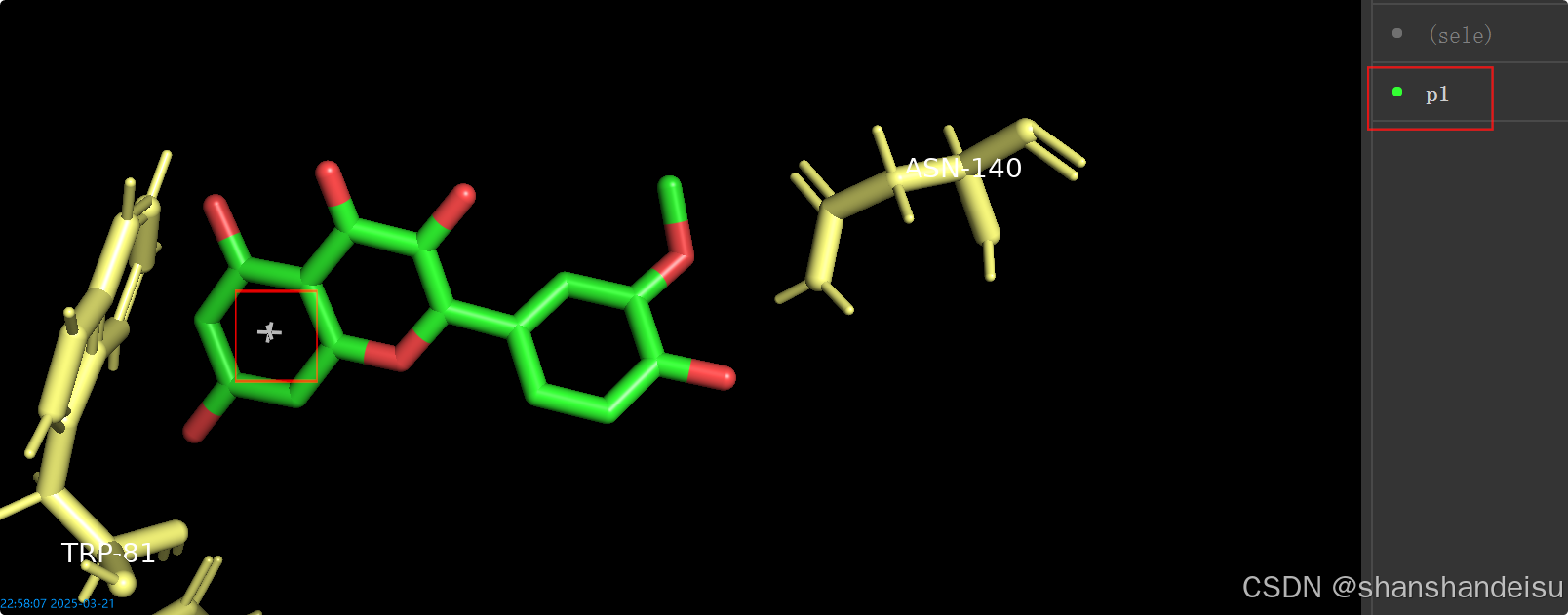
之后选中p1条目,将样式改为show/as/nb_spheres,然后可以看到p1中间的标志变为了球形。
然后在导航栏的settings/edit all,输入nonbonded_transparency,找到后改为0.9。
就能看到如下,圆球变的很透明了。
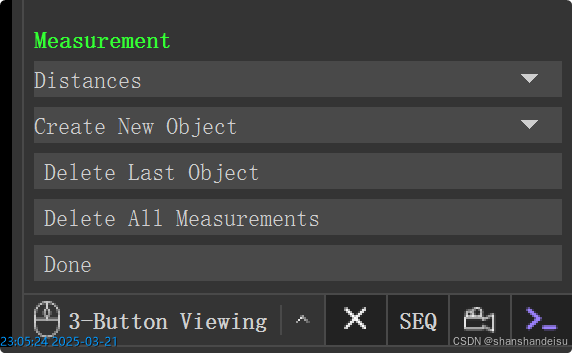
选中导航栏的wizard/measurement,看到右下角出现如下,说明我们进行测量模式。
根据之前的2D图像,来测量。
在测量前,再次在命令行输入以下命令,隐藏多余的水分子。
hide sticks, h.
如果中间你不小心连错了,可以点击对应的measure条目删除,或者直接点击delete last object。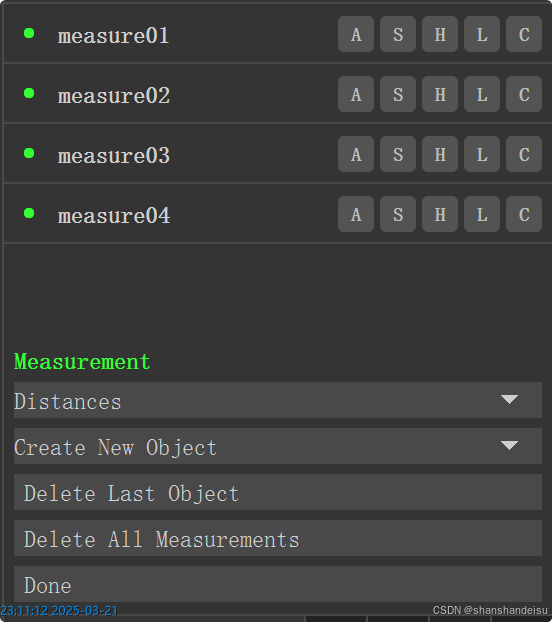
同时,将Π键对应的条目改为绿色,将氢键对应的条目改为紫色,保持和2D图的一致。最终如下。
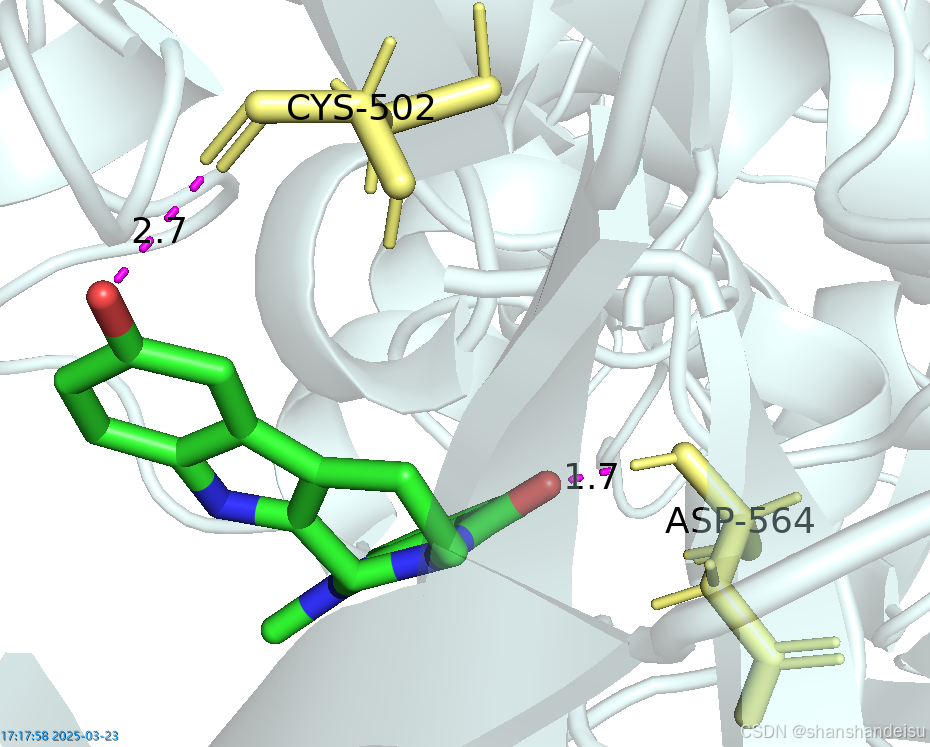
连完后点击done退出测量模式。
如果你是作用在苯环中心的键,你需要选中苯环中间的小圆点,然后选中与他相连的残基,它会自动进行测量:
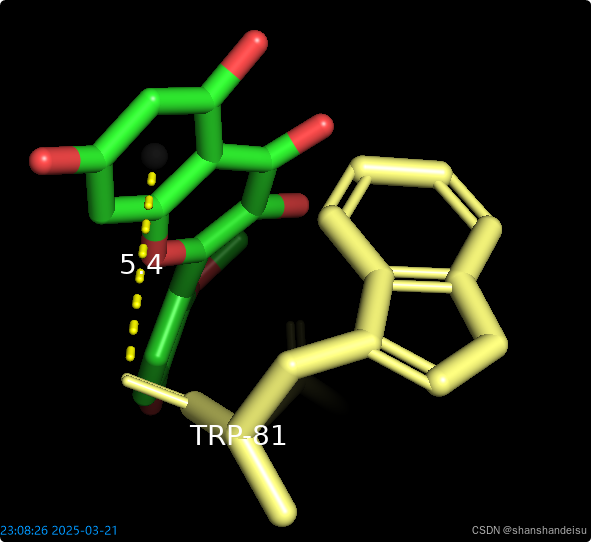
3.3.3.调整和输出
最后,在命令行输入:
zoom res or lig
将画面以配体和作用残基为中心,稍加旋转调整后如下:
将模式从view改为edit并按住ctrl,从而调整标签。
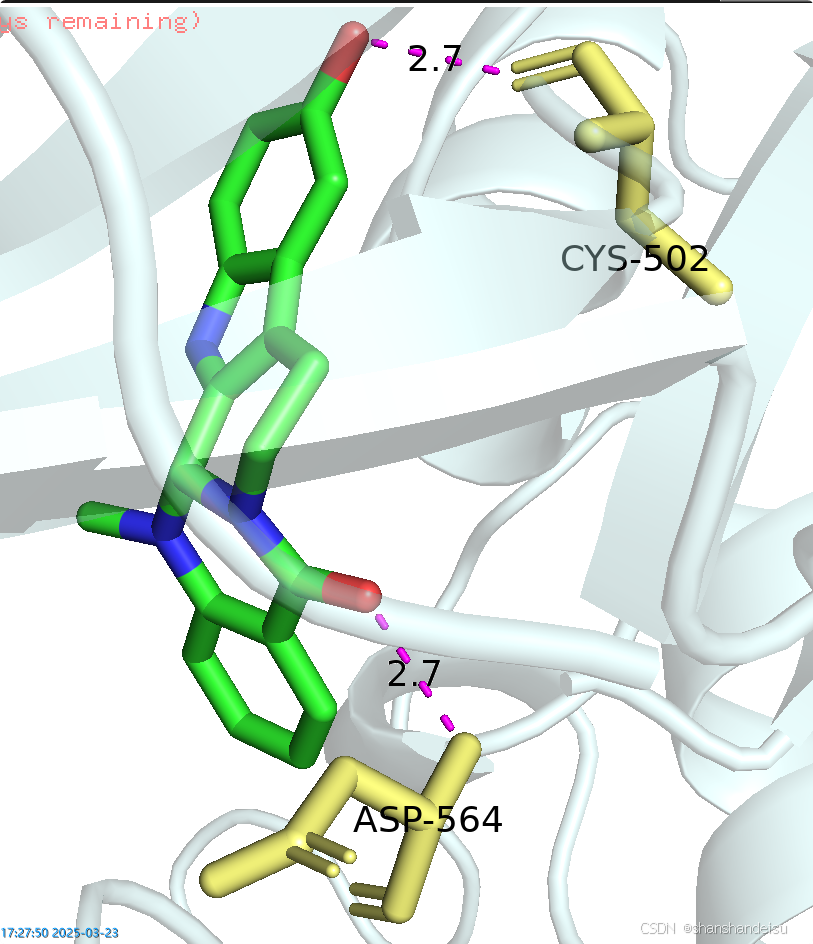
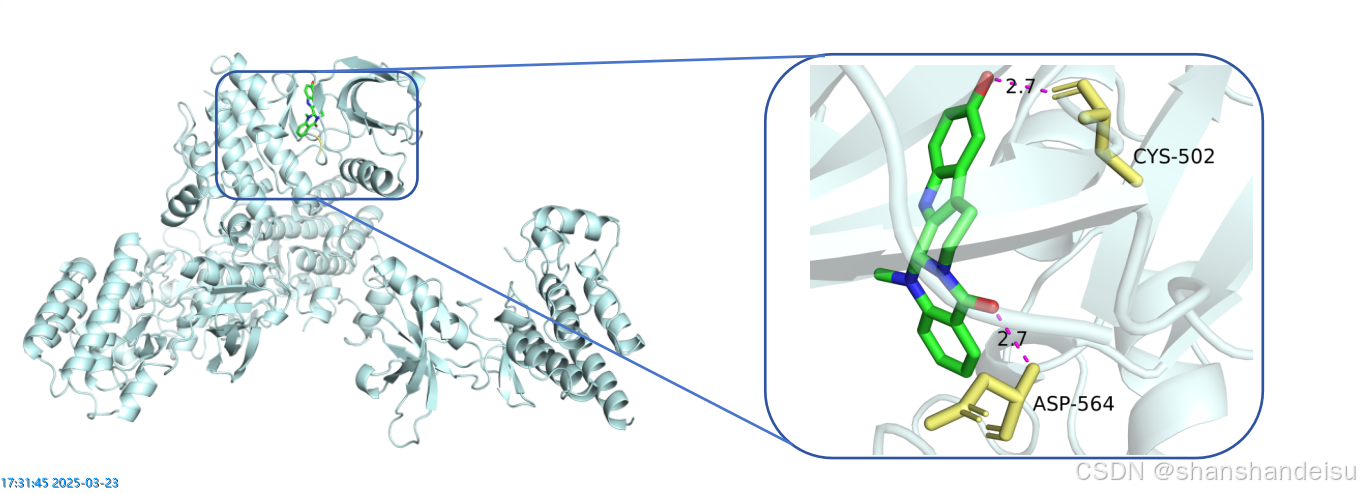
最后我们将两张图片导入word,裁剪掉空白的地方,调整成差不多大小,放到同一行里,插入圆角矩形的形状和线条,最终如下:



















 8616
8616

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











