



MolSnapper,一种通过整合专家知识为 SBDD 进行条件化的扩散模型。以 MolDiff 作为基础,在不重新训练 MolDiff 的情况下,对 MolDiff 的生成过程添加药效团、原子位置和类型等限制,以达到生成的分子能符合特定口袋的药效团的目的。
一、背景介绍
MolSnapper 来源于牛津大学统计系的 Charlotte M. Deane 教授为通讯作者的文章:《MolSnapper: Conditioning Diffusion for Structure Based Drug Design》。文章链接:MolSnapper: Conditioning Diffusion for Structure Based Drug Design | bioRxiv。该文章于 2024 年 3 月 30 日发表在 bioRxiv 上。

生成能够有效结合到预期靶标蛋白口袋中的分子仍然是当前基于结构的分子生成领域的挑战之一。控制设计/生成过程并结合先验知识将极大有助于生成适合特定结合位点的分子。MolSnapper 是一种能够通过整合 3D 药效团等专家知识来为基于结构的药物设计进行条件化扩散的新工具。在 CrossDocked 和 Binding MOAD 数据集上的测试结果表明,MolSnapper 能够生成更适合给定结合位点的分子,在结构和化学相似性上与原始分子高度一致。相较于没有口袋条件限制的 MolDiff 和 SILVR,MolSnapper 能够生成接近/超过两倍的有效分子。
二、模型介绍
在基于结构的药物设计(SBDD)领域,设计能够精准结合靶蛋白的配体依然是一个持续的挑战。相较于自回归模型由于顺序生成累积的误差和无法充分捕捉现实场景的限制,扩散模型能够同时建模原子之间的局部和全局相互作用。但由于蛋白质-配体复合物数据的有限,模型往往学到的是数据集的偏差,而不是掌握配体-蛋白质相互作用背后的真正生物物理原理。通过在生成阶段使用药效团约束进行控制的策略,可以显著提高生成分子的质量。
条件化是一种在生成过程中为模型提供额外信息或约束的方法,可以用于实现对生成分子的控制。预训练的扩散模型是一种实现这种控制的方法。例如 SILVR 方法在去噪过程中引入了一个精炼步骤,使用生成的配体和加入噪声的参考分子的线性组合,以生成能够适配结合位点的新分子,但该方法要求生成分子和参考分子有相同数量的原子,并且由于缺少对蛋白质的学习,生成许多一些和蛋白质冲突的分子。
另一种条件化方法是 DiffSBDD 使用的 inpainting。在该策略中,一个无条件的扩散模型可以通过在采样过程中修改概率转移步骤,将上下文信息注入采样过程,从而生成近似的条件样本。DiffSBDD 直接将蛋白质信息整合到模型中(而不是在条件化过程中),但它在训练中仍然局限于仅使用蛋白质-配体复合物数据。
当前基于结构的分子生成方法生成的分子常常不符合物理有效性,并且也难以匹配和超越真实配体与蛋白质靶标之间的相互作用。
针对上述提到的问题,作者提出了 MolSnapper,一种为 SBDD (structure-based drug design)设计的,通过整合专家知识的条件扩散模型。MolSnapper 不仅可以生成合理有效的分子,还能够形成类似于真实配体与蛋白质之间的氢键或其他相互作用。MolSnapper 利用了 3D 药效团形式的结构信息,此外,还可以根据特定的靶标蛋白选择合适的约束类型。
在 CrossDocked 和 Binding MOAD 数据集上的评估结果表明,MolSnapper 在生成与参考分子相似度较高的分子方面优于 SILVR 方法,可以比肩使用相互作用作为条件训练的分子生成模型,并且成功再现了参考配体与蛋白质之间的绝大多数氢键相互作用,生成有效分子的数量约为其他方法的两倍。
2.1 模型框架
MolSnapper 利用了药物分子空间中的大量可用训练数据,并通过使用 3D 药效团将物理上有意义的 3D 结构信息整合到生成过程中。作者选择 MolDiff 作为基础模型,预先在 GEOM-Drug 数据集上进行了训练。MolSnapper 使用的是条件化的策略是在不改变模型权重的情况下进行的。MolSnapper 从参考分子中提取的 3D 药效团包括氢键供体和受体,利用这些药效团的3D位置和类型(供体或受体)对MolDiff 的反向(去噪)生成过程进行条件化。
为了在反向(去噪)生成过程中约束原子位置,作者引入了一个掩码(用于标记哪些原子可以移动,哪些原子不可以移动),用于限制药效团位的修改。在每个阶段,模型强制某些原子位置接近固定位置。在初期步骤中,允许原子稍微向固定位置移动,而在最后的步骤中,则将它们牢牢固定在适当的位置。与其他方法不同的是,MolSnapper 采用了使用药效团参考点原始位置的策略,而不引入额外的噪声。
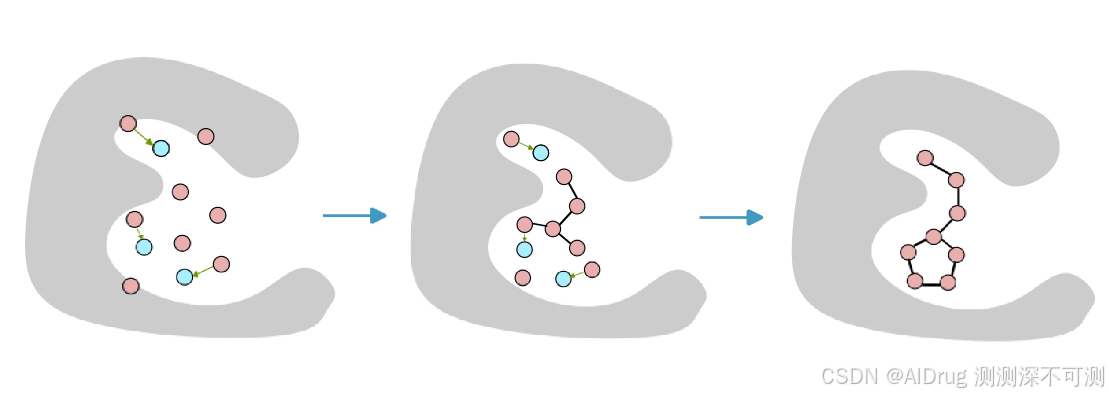
下图展示了受固定位置约束的原子位置采样过程。粉色球体表示生成分子的原子位置,蓝色球体表示固定参考点的位置,灰色网格代表蛋白质表面。在扩散过程输出时间点 t 的位置后,每一步中某些原子逐步向固定位置移动,帮助细化分子结构。
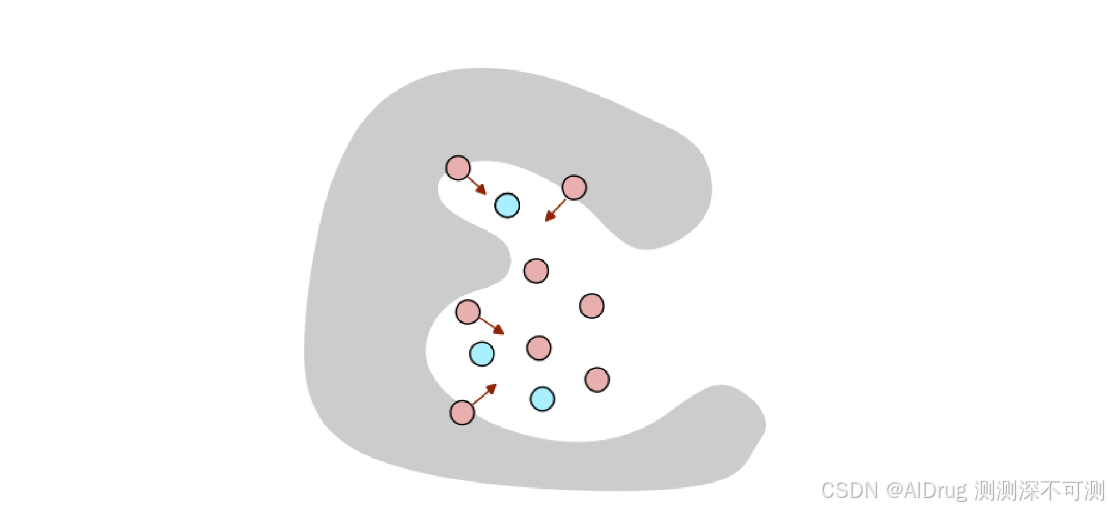
由于 MolDiff 模型未使用蛋白质数据进行训练,因此它无法处理结合口袋的相关信息。因此,必须引入引导机制,以防止生成的配体与蛋白质之间发生碰撞或距离过近。下图展示了分子生成过程中的冲突引导。蓝色表示固定的位置,粉色表示生成分子的原子位置,灰色网格代表蛋白质表面。冲突引导机制用于防止生成的配体与蛋白质之间发生冲突或距离过近。冲突引导损失的梯度为增强分子生成提供了优化方向。
为了提高生成分子的可解释性和控制性,作者对原子类型施加了约束。具体来说,作者规定氮(N)作为氢键供体的原子,氧(O)作为氢键受体的原子。在整个迭代采样过程中,始终将 N 作为供体和 O 作为受体的概率设置为 1 。模型中没有明确处理氢原子,这种方法为领域专家提供了灵活性,以便在最终输出中根据需要修改这个设置。
2.2 数据集和基线模型
作者在人工对接的 CrossDocked 2020 数据集和实验的 Binding MOAD 结构数据集上评估了条件化方法的有效性。
对于 CrossDocked 2020 数据集,仅保留了高质量的对接姿态(RMSD < 1Å)和多样化的蛋白质(使用 MMseqs2 计算的序列相似性 < 30%)。训练集为 100,000 对高质量的蛋白质-配体对,测试集为 100 对。通过 PoseBusters 对测试集进行评估,并使用 Open Drug Discovery Toolkit (ODDT) 检查配体与蛋白之间是否存在氢键相互作用。为了确保测试集的高质量,只保留通过所有 PoseBusters 测试的数据,并确保每个测试案例都包含相关的药效团(仅选择氢键相互作用数目超过3个的数据,H 键的数量有点多)。测试集最终为筛选出的 73 个复合物。
Binding MOAD 是一个实验确定的蛋白-配体复合物数据集。基于蛋白的酶委员会编号进行过滤和分类。40,344 个蛋白-配体对用于训练,并从中选取了 130 个用于测试。与CrossDocked 2020 数据集类似,对测试集的选择标准是通过所有 PoseBusters 测试,并且具有超过3个氢键相互作用。最终从 130 个复合物中筛选出了 43 个复合物。
作者将 MolSnapper 与 MolDiff 、 SILVR 和 DiffSBDD 在蛋白口袋中生成配体的方法进行了比较。
为了验证 MolSnapper 条件化的有效性,将其与未进行条件化的 MolDiff 进行比较。对于MolDiff 生成的配体,因为 MolDiff 生成的分子并未在口袋内作为条件生成,因此需要使用 Vina 对其进行对接。
SILVR 是一种为适配蛋白质结合位点生成分子而设计的方法。作者使用了他们的代码,指定药效团的位置和原子类型,并将剩余原子视为虚拟原子。
DiffSBDD 采用了一种用于灵活分子设计的条件化方法,称为 Inpainting 。对于 DiffSBDD,作者提供了药效团位置和原子类型作为固定原子的输入。
2.3 评价指标
(Shape and color similarity score)评估生成的分子与参考分子的3D相似性。范围从 0(不匹配)到 1(完全匹配)。颜色相似性功能评估药效团特征的重叠,而形状相似性则通过两种构象体之间的简单体积比较来衡量。
SA(Synthetic Accessibility)衡量化合物的合成难易程度。它评估分子的复杂性,并利用已合成化学品的历史数据来估计合成难度。通常取值范围为 1 - 10 ,数值越大合成难度越大,但作者为和其他指标保持一致做了标准化处理,取值范围变成 0 - 1,数值越高表明更容易合成。
Same ODDT Interaction 计算生成的配体与蛋白质之间的氢键,与参考配体和蛋白质之间的氢键的相似度百分比,使用的是Open Drug Discovery Toolkit (ODDT)。
PoseBusters Pass Rate 表示通过 PoseBusters 测试的生成分子的百分比。PoseBusters 测试套件检查生成的化学构象的化学有效性和一致性、分子内有效性以及分子间有效性,评估生成构象的物理化学一致性和结构合理性。
2.4 模型性能
2.4.1 非口袋训练条件下的对比
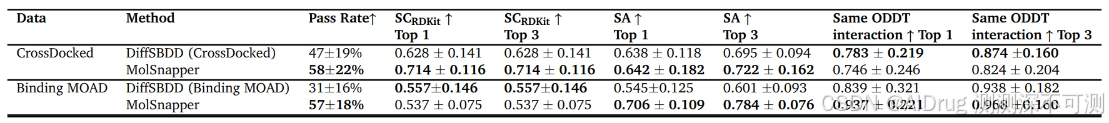
下表展示了在 CrossDocked 数据集上,MolSnapper 与没有任何条件方法的 MolDiff 和 SILVR 的比较结果。SILVR 仅在分子数据集上训练,而不是在蛋白质-分子数据上训练 。MolDiff(无条件基础模型)生成了具有较高 SA 评分的配体(Top 1 的平均值为 0.866)。为了检查这些分子如何与蛋白质相互作用,将它们对接到蛋白质口袋中。这些对接的配体倾向于通过 PoseBusters 测试(91% 的 PoseBusters 通过率)。然而,MolDiff 生成的配体通常无法重现参考配体所显示的相互作用(Top 1 的平均 Same ODDT Interaction 为 0.276)。
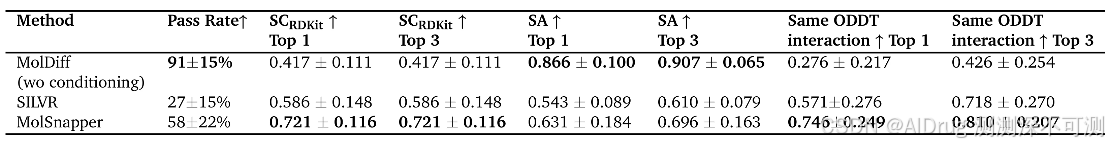
SILVR 和 MolSnapper 生成的配体基于 3D 药效团位置进行条件化,与非条件的 MolDiff 相比,这两种方法生成的分子平均 SA 评分较低,其中 MolSnapper 生成的分子在合成方面更好一些(SILVR 的Top 1 平均 SA 为 0.543,而 MolSnapper 为 0.631)。尽管 SILVR 和 MolSnapper 的 PoseBusters 通过率低于原始的 MolDiff,但 MolSnapper 有 58% 的分子通过 PoseBusters 测试,而 SILVR 仅有 27% 。
MolSnapper 生成分子的(Top 1 的平均值为 0.721)显著高于 MolDiff 或 SILVR 生成的分子(分别为0.417和0.586)。此外,SILVR 和 MolSnapper 在再现原始氢键方面均优于 MolDiff 。MolSnapper 在 Same ODDT Interaction 指标中的 Top 1 平均得分为 0.746,优于 SILVR 的 0.571。这些结果表明MolSnapper 在相互作用指标上优于 SILVR 。
性能差异可以归因于 SILVR 生成分子时观察到的多个问题。一个问题是 SILVR 生成的分子中存在大量不连通的片段。在评估中,作者通过选择生成的最大片段来解决这一问题。此外,由于SILVR 的模型未整合蛋白质信息,生成分子与蛋白质存在冲突。此外,基础模型 MolDiff 优于SILVR 使用的基础模型 EDM,因为它对分子的键进行建模和扩散,从而生成更具有效性和合成可及性的分子。
下图展示了 SILVR 和 MolSnapper 方法生成的分子示例,重点关注 得分最高的配体。在第一行中,SILVR 未能生成满足给定药效团约束的分子,生成的小片段无法与蛋白质形成氢键。在第二行中,生成了适当大小的分子,但未能形成与参考配体相同的键。
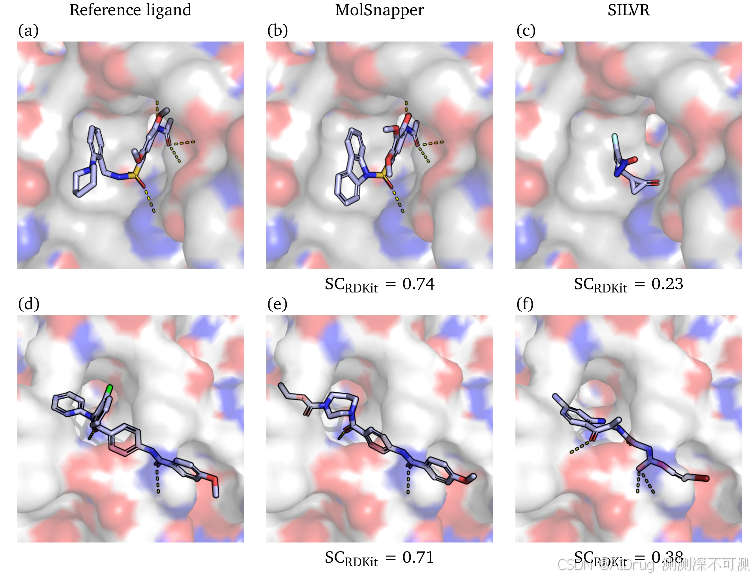
在不进行重新对接的情况下,MolSnapper 达到了 58% 的 PoseBusters 通过率,与原始配体高度相似,并能再现大多数现有的氢键相互作用。这些方面的重要性在于保留了关键的相互作用和与真实配体的相似性,这是保持生成分子在药理学相关性和有效性方面至关重要的因素。
2.4.2 有口袋条件下训练的对比
作者将 MolSnapper 与 DiffSBDD 进行了基准测试,DiffSBDD 是一种专门在蛋白质-配体复合物数据上训练的 3D 条件扩散模型。下表比较了在 CrossDocked 数据集上训练的 DiffSBDD 与 MolSnapper,以及在 Binding MOAD 数据集上 训练的DiffSBDD 与 MolSnapper 的性能。DiffSBDD 和 MolSnapper 在生成与原始分子相似的分子()和再现结合相互作用( Same ODDT Interaction)方面表现相当,但 MolSnapper 生成的分子在物理可行性(PoseBusters 通过率)和合成可及性(SA 评分)方面表现更好。这些结果表明,基于更大分子空间进行训练并针对口袋生成进行条件化,而不仅仅在蛋白质-分子集合上进行训练,能更好地代表实际分子。
2.4.3 文章案例对比
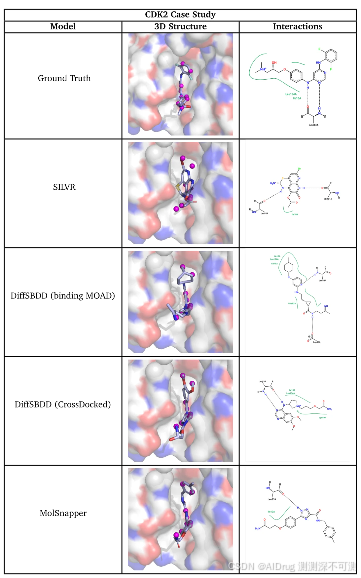
接下来,作者比较了一个针对广泛研究的治疗靶点 CDK2(PDB ID: 1h00)的案例研究。作者将研究范围扩展到氢键之外,考虑了 PLIP 提取的所有相互作用,这些相互作用包括氢键和疏水相互作用。特别选择了参与这些相互作用的原子作为参考点。
作者使用 SILVR、DiffSBDD(使用 Binding MOAD 训练)、DiffSBDD(使用 CrossDocked 训练)和 MolSnapper 分别生成 100 个分子。结果显示,使用 Binding MOAD 训练的 DiffSBDD 在配体相似性和再现结合相互作用方面表现最佳,获得了最高的 为 0.582(SILVR、DiffSBDD(CrossDocked)和 MolSnapper 分别为 0.327、0.569、0.574)和最高的相同 PLIP 相互作用得分为 0.7(SILVR、DiffSBDD(CrossDocked)和 MolSnapper 分别为 0.2、0.6、0.5)。MolSnapper 在 PoseBuster 通过率上表现最佳,为 69%(SILVR、DiffSBDD(CrossDocked)和 DiffSBDD(Binding MOAD)分别为 15%、57%、45%),并且生成了更多接近真实配体的分子。其
> 0.5 的分子比例为 12% (SILVR、DiffSBDD(CrossDocked)和DiffSBDD(Binding MOAD)分别为 0%、8%、4%)。下图比较了 CDK2 案例研究中的 3D 结构和相互作用,展示了所有方法中
> 0.5 的最佳结果分子。
2.4.4 MolSnapper 消融研究
为探究初始化和参考条件化对生成的影响,作者在 CrossDocked 数据集上比较了三种初始化策略和参考点处理方式。
(1)药效团固定点附近的随机初始化,参考位置固定:生成分子的初始位置为药效团固定点附近的随机噪声,且参考位置在扩散逆过程期间保持固定。
(2)随机初始化,参考位置固定:生成分子的初始位置为随机噪声,参考位置在整个生成过程中保持固定,类似于前一种情况。
(3)药效团固定点附近的随机初始化,参考点带噪声:生成分子的初始位置为药效团固定点附近的随机噪声,参考点在扩散逆过程期间基于预定义的噪声缩放计划被添加噪声。
下表总结了消融研究结果,重点是噪声对使用 GNN 进行分子生成的影响。最佳的 分数是在初始化 1 中获得的,即药效团固定点附近的随机初始化,但不对参考点添加噪声。这表明,稳定的参考点在保持生成过程中关键的配体-靶点相互作用方面非常重要。

三、MolSnapper 评测
3.1 安装环境
复制代码项目:
git clone https://github.com/oxpig/MolSnapper.git通过项目提供的 env.yml 创建 MolSnapper 环境
conda env create -f env.yml
conda activate MolSnapper3.2 分子生成案例测试
项目提供训练好的 MolDiff 模型,保存在谷歌网盘中,链接是:https://drive.google.com/drive/folders/1zTrjVehEGTP7sN3DB5jaaUuMJ6Ah0-ps。(注:MolDiff 模型也作为 MolSnapper 的基础模型,MolSnapper 直接使用 MolDiff 的 checkpoint)网盘中的内容如下:
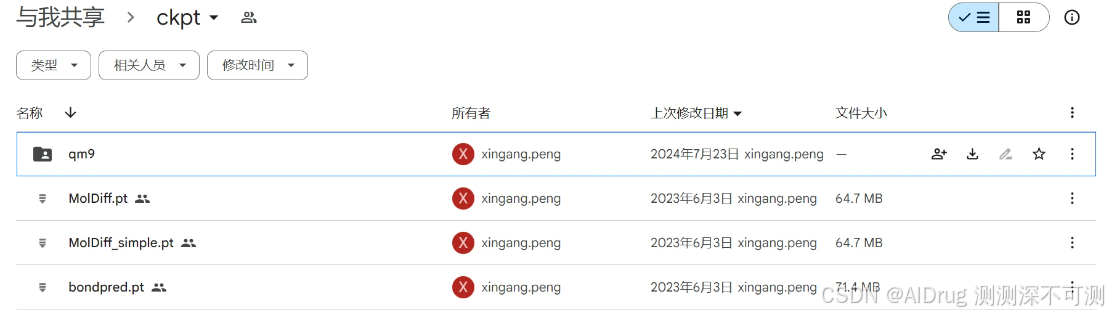
MolDiff.pt 是预训练的完整版本的 MolDiff 模型,具备生成和推断化学分子结构的能力。这个模型能够处理复杂的分子生成任务,包括分子中原子的空间布局和键的生成。
MolDiff_simple.pt 是 MolDiff 模型的简化版本,在训练过程中没有使用新的“键噪声调度”(即调整生成过程中引入的噪声与分子键相关的步骤)。虽然它仍然能够生成分子,但在处理分子键的精确度方面可能不如完整模型。
bond_predictor.pt:预训练的键预测器,是一个独立的键预测器模型,专门用于在生成分子时预测分子内各个原子之间的键,并在采样过程中引导生成过程,使生成的分子结构更合理、化学上更稳定。
网盘中训练好的模型下载后放在 ./ckpt 文件夹中,目录结构如下:
.
|-- MolDiff.pt
|-- MolDiff_simple.pt
|-- README.md
`-- bondpred.pt
0 directories, 4 files
接下来的项目内置案例和自定义案例测试,均使用与 MolDiff 相同的配置文件,随机种子设置为 2023,每个案例生成 100 个分子。生成分子配置文件保存在 ./configs/sample/sample_MolDiff.yml 。具体配置内容如下:
model:
checkpoint: ckpt/MolDiff.pt
bond_predictor: ckpt/bondpred.pt
sample:
seed: 2023
batch_size: 512
num_mols: 100
save_traj_prob: 0.02
guidance:
- uncertainty
- 1.e-43.2.1 内置案例
项目中提供的测试案例是 CDK2 (PDB ID: 1h00)。创建 ./data/example_1h00 文件夹,从 PDB 数据库下载的蛋白结构 1h00.pdb 放在 ./data/example_1h00 文件夹中。蛋白口袋中的 FCP 配体作为参考分子,配体在口袋中的构象如下:
口袋中分子的 2D 结构,如下:
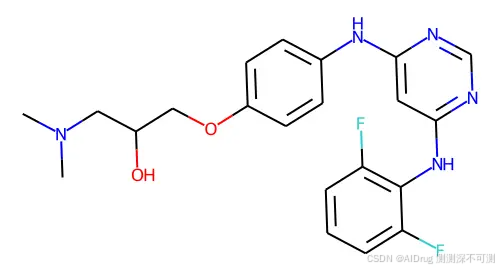
1.结构预处理
在进行分子生成前,需要处理原始蛋白-配体复合物结构,删除氢原子,溶剂和离子等,分别保存蛋白结构和配体结构,命令如下:
python ./scripts/clean_and_split.py \
--in-dir ./data/example_1h00 \
--proteins-dir ./data/example_1h00 \
--ligands-dir ./data/example_1h00--in-dir ./data/example_1h00 指定原始结构文件 1h00.pdb 所在的位置。--proteins-dir ./data/example_1h00 和 --ligands-dir ./data/example_1h00 指定处理后的蛋白和配体的保存位置。
命令报错如下:
/bin/sh: 1: pdb_selmodel: not found
/bin/sh: 1: pdb_delelem: not found
/bin/sh: 1: pdb_delhetatm: not found
/bin/sh: 1: pdb_selhetatm: not found
/bin/sh: 1: pdb_delelem: not found
/bin/sh: 1: pdb_delelem: not found
Problem getting relevant ligands PDB=3wze: Python argument types in
rdkit.Chem.rdmolops.GetMolFrags(NoneType)
did not match C++ signature:
GetMolFrags(RDKit::ROMol mol, bool asMols=False, bool sanitizeFrags=True, boost::python::api::object frags=None, boost::python::api::object fragsMolAtomMapping=None)
根据报错显示,环境中缺少 pdb-tools ,安装后预处理命令可以正常执行,安装命令如下:
pip install pdb-tools原始结构处理后,蛋白和配体结构分别保存,./data/example_1h00 的目录如下。其中,1h00.pdb 是复合物结构, 1h00_0.sdf 和 1h00_protein.pdb 是分别保存的配体和蛋白。
.
|-- 1h00.pdb
|-- 1h00_0.sdf
`-- 1h00_protein.pdb
0 directories, 3 files2.数据准备
接着,根据处理好的蛋白口袋和配体提取模型的输入信息,具体命令如下:
python scripts/prepare_single_complex.py \
--root_dir ./data/example_1h00 \
--ligand_filename 1h00_0.sdf \
--protein_filename 1h00_protein.pdb \
--out_pockets_path ./data/example_1h00/processed_pocket_1h00.pkl--root_dir ./data/example_1h00 指定蛋白结构和配体结构的目录;--ligand_filename 1h00_0.sdf 和 --protein_filename 1h00_protein.pdb 分别蛋白结构和配体结构的文件名;--out_pockets_path ./data/example_1h00/processed_pocket_1h00.pkl 指定处理后的 pkl 格式的口袋信息文件。
命令输出的蛋白口袋文件在 ./data/example_1h00/processed_pocket_1h00.pkl,包含口袋的全部(full)原子坐标和类型以及主链(backbone)的原子坐标和类型。数据结构示意如下:
{
'full_coord': pocket_coords_full,
'full_types': pocket_types_full,
'bb_coord': pocket_coords_bb,
'bb_types': pocket_types_bb,
}3.分子采样
运行命令如下:
python scripts/sample_single_pocket.py \
--outdir ./output_1h00 \
--config ./configs/sample/sample_MolDiff.yml \
--batch_size 32 \
--pocket_path ./data/example_1h00/processed_pocket_1h00.pkl \
--sdf_path ./data/example_1h00/1h00_0.sdf \
--use_pharma True \
--clash_rate 0.1--outdir ./output_1h00 指定生成分子的保存路径。--pocket_path ./data/example_1h00/processed_pocket_1h00.pkl 输入数据处理好的蛋白口袋信息。--sdf_path ./data/example_1h00/1h00_0.sdf 指定配体结构。--use_pharma True 设置为 True,模型根据配体结构找到药效团位置,并作为分子生成的参考,尽可能保证生成分子和参考分子的相互作用不会相差太大。--clash_rate 0.1 表示控制分子生成过程中避免原子冲突的强度值。
生成命令输出:
[2024-09-08 07:21:34,090::sample::INFO] Namespace(config='./configs/sample/sample_MolDiff.yml', outdir='./output_1h00', pocket_path='./data/example_1h00/processed_pocket_1h00.pkl', sdf_path='./data/example_1h00/1h00_0.sdf', device='cuda:0', batch_size=32, mol_size=None, use_pharma=True, pharma_th=0.5, clash_rate=0.1, distance_th=1.0)
[2024-09-08 07:21:34,090::sample::INFO] {'model': {'checkpoint': 'ckpt/MolDiff.pt'}, 'bond_predictor': 'ckpt/bondpred.pt', 'sample': {'seed': 2023, 'batch_size': 512, 'num_mols': 100, 'save_traj_prob': 0.02, 'guidance': ['uncertainty', 0.0001]}}
[2024-09-08 07:21:34,090::sample::INFO] Loading data placeholder...
[2024-09-08 07:21:34,104::sample::INFO] Loading diffusion model...
[2024-09-08 07:21:34,222::sample::INFO] Building bond predictor...
[2024-09-08 07:23:11,082::sample::WARNING] Incomplete molecule: CCOCC(O)CN.CN1C(=O)NC2NNC(N)NC2C1=O
[2024-09-08 07:23:11,089::sample::WARNING] Incomplete molecule: CCN1C(=O)C2C(NCCCOCCO)NNC2N(C)C1=O.N
[2024-09-08 07:23:11,101::sample::INFO] Success: N#Cc1cccc(OCC=CC=NNC2NNC(N(CC(=O)O)C(C=O)C(=O)O)N2)c1O
...
[2024-09-08 07:42:49,209::sample::WARNING] Incomplete molecule: CCCCNC1C(C#N)C(N)NC(N2CCOCC2)N1O.NCC(O)CO
[2024-09-08 07:42:49,218::sample::WARNING] Incomplete molecule: C.NC=[SH]NC1NC(NO)C([N+](=O)O)N1.NN
[2024-09-08 07:42:49,229::sample::WARNING] Incomplete molecule: CCNC1NC2NCN(C=O)C(c3cccs3)N2N1.N.OCC(O)CO
[2024-09-08 07:42:49,231::sample::INFO] [Pool] Finished 60 | Failed 324
[2024-09-08 07:42:49,232::sample::INFO] Too many failed molecules. Stop sampling.当batch_size 设置为 32 时显存占用为 8 G, 设置生成 100 个分子,由于采样失败的不完整分子达到了 324 个,超过了 3 倍的生成分子数目,程序终止了采样,最后保存了 60 个分子。花费时间约 22 分钟。命令结果输出为两个文件夹,保存在 ./output_1h00 中,其目录如下:
.
|-- sample_MolDiff_20240908_072134_clash_rate_0.1
`-- sample_MolDiff_20240908_072134_clash_rate_0.1_SDF
2 directories, 0 files其中,sample_MolDiff_20240908_072134_clash_rate_0.1 文件夹中保存命令输出的记录文件、配置文件和生成分子的 SMILES 等信息。sample_MolDiff_20240908_072134_clash_rate_0.1_SDF 文件夹中保存每个生成分子的 SDF 格式文件。
通过 PoseBusters 过滤生成分子,评价基础的分子属性、计算与参考分子的 3D 相似度,使用 ODDT 计算生成分子和参考分子的氢键数目。具体命令如下:
python scripts/evaluate.py \
./output_1h00/sample_MolDiff_20240908_072134_clash_rate_0.1_SDF \
--protein_path ./data/example_1h00/1h00_protein.pdb \
--reflig_path ./data/example_1h00/1h00_0.sdf \
--save_path ./output_1h00/eval 命令第一个参数指定保存生成分子构象的文件夹;--protein_path 和 --reflig_path 分别指定蛋白结构和参考分子的位置; --save_path 指定评估结果保存的位置。
分子评估命令运行输出:
Namespace(gen_root='./output_1h00/sample_MolDiff_20240908_072134_clash_rate_0.1_SDF', protein_path='./data/example_1h00/1h00_protein.pdb', reflig_path='./data/example_1h00/1h00_0.sdf', save_path='./output_1h00/eval')
QED: Mean: 0.205 Median: 0.184 Std: 0.110
SA: Mean: 0.602 Median: 0.590 Std: 0.083
SuCOS_sim: Mean: 0.586 Median: 0.583 Std: 0.065
interaction: Mean: 2.067 Median: 2.000 Std: 1.031生成分子的平均 QED 和 SA 分别为 0.205 和 0.602。生成分子的类药性较低,可合成性良好。脚本输出的 SuCOS_sim 即 SC_{RDKit}均值为 0.586,生成分子和参考分子的药效团和形状基本匹配,相似性较高。interaction 均值为 2 表示生成分子和蛋白口袋平均形成两个氢键相互作用。
所有生成的分子如下:
MolSnapper_1h00
3.2.2 自定义案例
我们使用 3wze 的蛋白作为自己的测试案例。创建 ./data/example_3wze 文件夹,从 PDB 数据库下载的蛋白结构 3wze.pdb 放在文件夹中。配体在口袋中的构象如下:
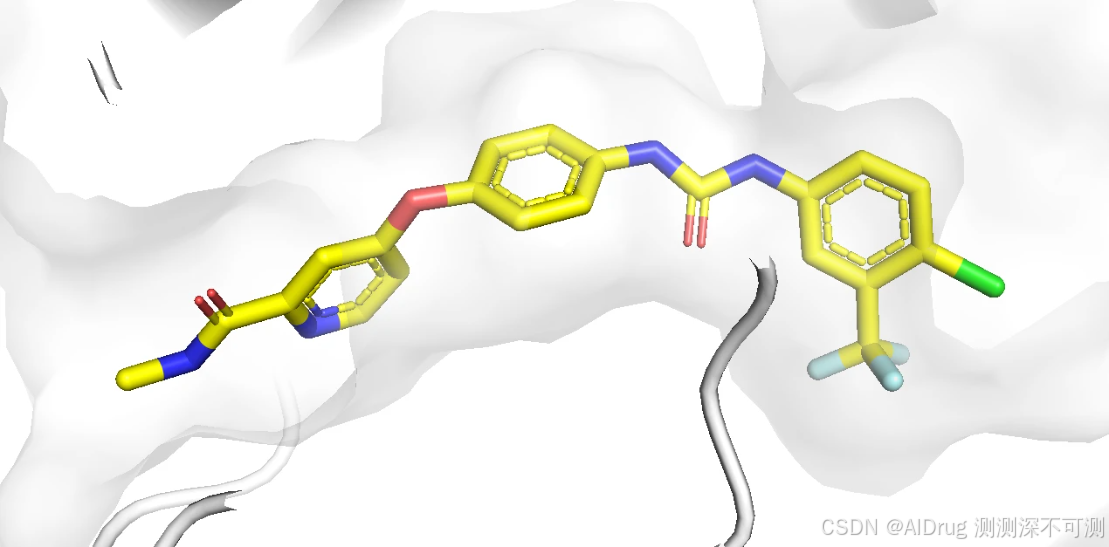
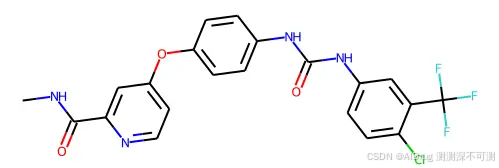
口袋中分子的 2D 结构,如下:
首先,处理原始蛋白-配体复合物结构,删除氢原子,溶剂和离子等,分别保存蛋白结构和配体结构,命令如下:
python ./scripts/clean_and_split.py \
--in-dir ./data/example_3wze \
--proteins-dir ./data/example_3wze \
--ligands-dir ./data/example_3wze --in-dir ./data/example_3wze 指定原始结构文件 3wze.pdb 所在的位置。--proteins-dir ./data/example_3wze 和 --ligands-dir ./data/example_3wze 指定处理后的蛋白和配体的保存位置。
原始结构处理后,蛋白和配体结构分别保存,./data/example_3wze 的目录如下。其中,3wze.pdb 是复合物结构, 3wze_0.sdf 和 3wze_protein.pdb 是分别保存的配体和蛋白。
.
|-- 3wze.pdb
|-- 3wze_0.sdf
`-- 3wze_protein.pdb
0 directories, 3 files接着,根据处理好的蛋白口袋和配体提取模型的输入信息,具体命令如下:
python scripts/prepare_single_complex.py \
--root_dir ./data/example_3wze \
--ligand_filename 3wze_0.sdf \
--protein_filename 3wze_protein.pdb \
--out_pockets_path ./data/example_3wze/processed_pocket_3wze.pkl--root_dir ./data/example_3wze 指定蛋白结构和配体结构的目录;--ligand_filename 3wze_0.sdf 和 --protein_filename 3wze_protein.pdb 分别蛋白结构和配体结构的文件名;--out_pockets_path ./data/example_3wze/processed_pocket_3wze.pkl 指定生成的口袋信息文件暴保存位置。
命令输出的蛋白口袋文件在 ./data/example_3wze/processed_pocket_3wze.pkl,包含口袋的全部(full)原子坐标和类型以及主链(backbone)的原子坐标和类型。数据结构示意如下:
{
'full_coord': pocket_coords_full,
'full_types': pocket_types_full,
'bb_coord': pocket_coords_bb,
'bb_types': pocket_types_bb,
}根据配置文件,基于蛋白口袋进行分子采样。命令如下:
python scripts/sample_single_pocket.py \
--outdir ./output_3wze \
--config ./configs/sample/sample_MolDiff.yml \
--batch_size 32 \
--pocket_path ./data/example_3wze/processed_pocket_3wze.pkl \
--sdf_path ./data/example_3wze/3wze_0.sdf \
--use_pharma True \
--clash_rate 0.1--outdir ./output_3wze 指定生成分子的保存路径。--pocket_path ./data/example_3wze/processed_pocket_3wze.pkl 输入数据处理好的蛋白口袋信息。--sdf_path ./data/example_3wze/3wze_0.sdf 指定配体结构。--use_pharma True 设置为 True,模型根据配体结构找到药效团位置,并作为分子生成的参考,尽可能保证生成分子和参考分子的相互作用不会相差太大。--clash_rate 0.1 表示控制分子生成过程中避免原子冲突的强度值。
生成命令输出:
[2024-09-08 09:46:55,376::sample::INFO] Namespace(config='./configs/sample/sample_MolDiff.yml', outdir='./output_3wze', pocket_path='./data/example_3wze/processed_pocket_3wze.pkl', sdf_path='./data/example_3wze/3wze_0.sdf', device='cuda:0', batch_size=32, mol_size=None, use_pharma=True, pharma_th=0.5, clash_rate=0.1, distance_th=1.0)
[2024-09-08 09:46:55,377::sample::INFO] {'model': {'checkpoint': 'ckpt/MolDiff.pt'}, 'bond_predictor': 'ckpt/bondpred.pt', 'sample': {'seed': 2023, 'batch_size': 512, 'num_mols': 100, 'save_traj_prob': 0.02, 'guidance': ['uncertainty', 0.0001]}}
[2024-09-08 09:46:55,377::sample::INFO] Loading data placeholder...
[2024-09-08 09:46:55,391::sample::INFO] Loading diffusion model...
[2024-09-08 09:46:55,609::sample::INFO] Building bond predictor...
[2024-09-08 09:48:37,093::sample::WARNING] Reconstruction error encountered.
[2024-09-08 09:48:37,101::sample::WARNING] Incomplete molecule: CC1=Nc2c(ccc3c2O3)C12c1nnnn12.Cc1noc(C#N)n1
[2024-09-08 09:48:37,110::sample::INFO] Success: NC(=O)c1cc(COc2ccc(C3NNNN3)cc2)on1
...
[2024-09-08 10:05:04,793::sample::INFO] Success: NC(=O)C=C(N)COc1ccc(N2NNN2)cc1
[2024-09-08 10:05:04,807::sample::INFO] Success: CC1NC(C=C(N)c2cc3cc(-c4nnn[nH]4)ccc3c3c2O3)O1
[2024-09-08 10:05:04,809::sample::INFO] [Pool] Finished 100 | Failed 122batch_size 设置为 32 显存为 8 G, 设置生成 100 个分子,保存了 100 个分子。花费时间约 20 分钟。命令结果输出为两个文件夹,保存在 ./output_3wze 中,目录如下:
.
|-- sample_MolDiff_20240908_094655_clash_rate_0.1
`-- sample_MolDiff_20240908_094655_clash_rate_0.1_SDF
2 directories, 0 files其中,sample_MolDiff_20240908_094655_clash_rate_0.1 文件夹中保存命令输出的记录文件、配置文件和生成分子的 SMILES 等信息。sample_MolDiff_20240908_094655_clash_rate_0.1_SDF 文件夹中保存每个生成分子的 SDF 格式文件。
通过 PoseBusters 过滤生成分子,评价基础的分子属性、计算与参考分子的 3D 相似度,使用 ODDT 计算生成分子和参考分子的氢键数目。具体命令如下:
python scripts/evaluate.py \
./output_3wze/sample_MolDiff_20240908_094655_clash_rate_0.1_SDF \
--protein_path ./data/example_3wze/3wze_protein.pdb \
--reflig_path ./data/example_3wze/3wze_0.sdf \
--save_path ./output_3wze/eval 命令第一个参数指定保存生成分子构象的文件夹;--protein_path 和 --reflig_path 分别指定蛋白结构和参考分子的位置;--save_path 指定评估结果保存的位置。
分子评估命令运行输出:
Namespace(gen_root='./output_3wze/sample_MolDiff_20240908_094655_clash_rate_0.1_SDF', protein_path='./data/example_3wze/3wze_protein.pdb', reflig_path='./data/example_3wze/3wze_0.sdf', save_path='./output_3wze/eval')
QED: Mean: 0.434 Median: 0.437 Std: 0.158
SA: Mean: 0.676 Median: 0.690 Std: 0.085
SuCOS_sim: Mean: 0.501 Median: 0.504 Std: 0.045
interaction: Mean: 4.320 Median: 4.000 Std: 1.529生成分子的平均 QED 和 SA 分别为 0.434 和 0.676。生成分子的类药性较低,可合成性良好。评估输出的 SuCOS_sim 即 SC_{RDKit}均值为 0.501,生成分子和参考分子的药效团和形状匹配程度一般,相似性中等。interaction 均值为 4.320 表示生成分子和蛋白口袋平均形成四个氢键相互作用。
所有生成的分子如下:
MolSnapper_3wze
qvina score 排名前 3 的分子的 2D 结构如下,对应的 qvina score 分数分别为 -12.4, -11.3, -11.1:
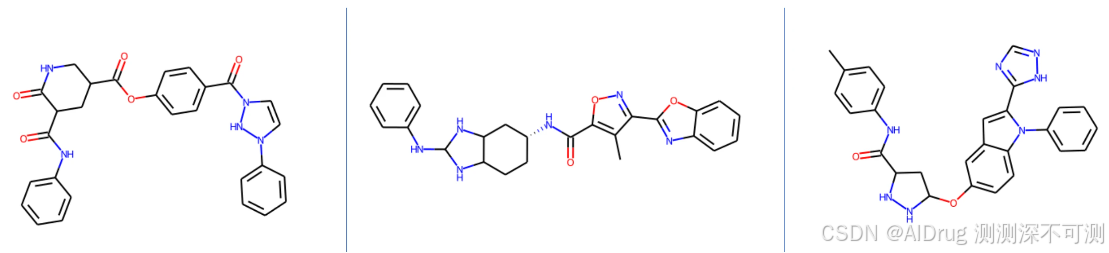
qvina score 排名前 3 的分子在口袋中的 Pose 如下:
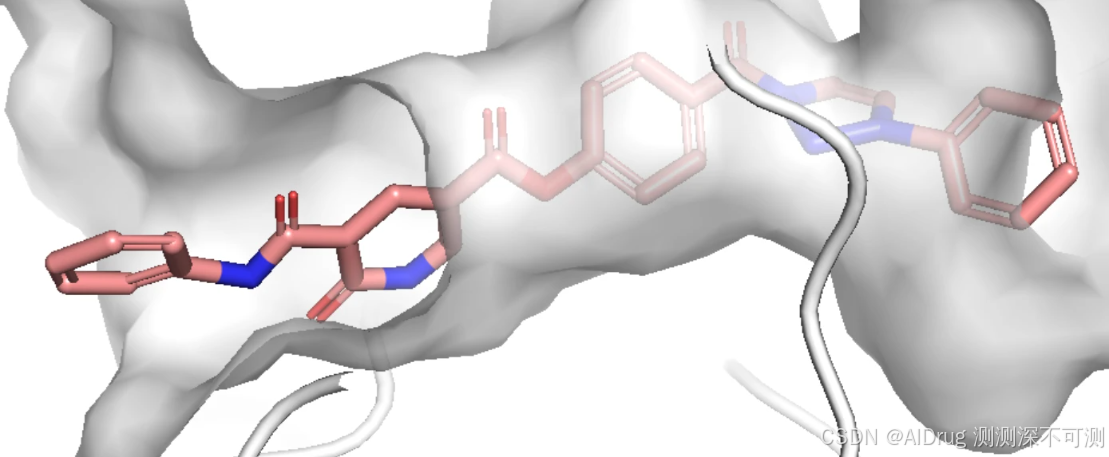
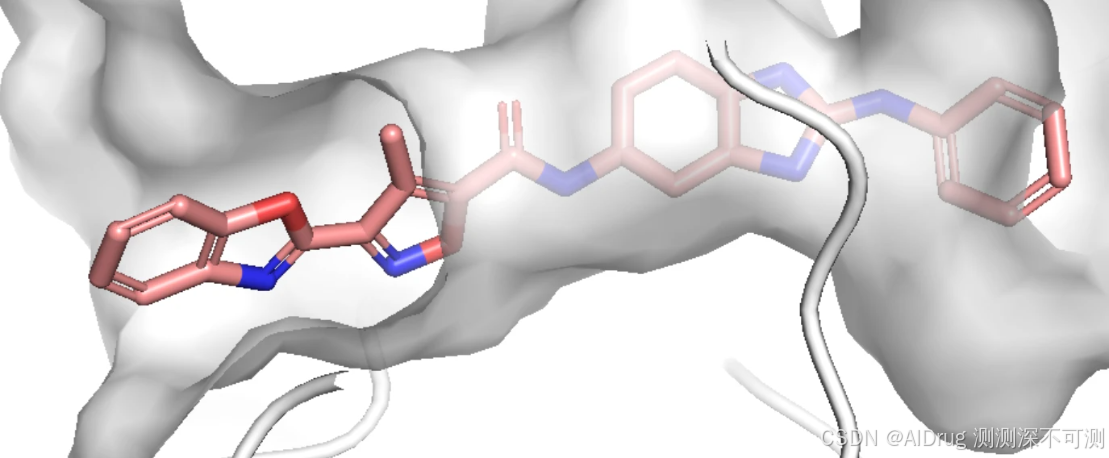
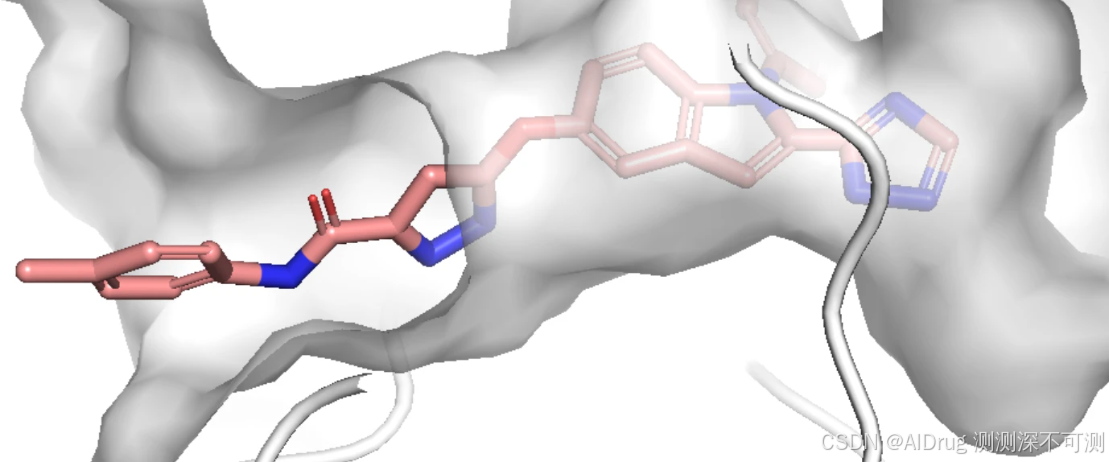
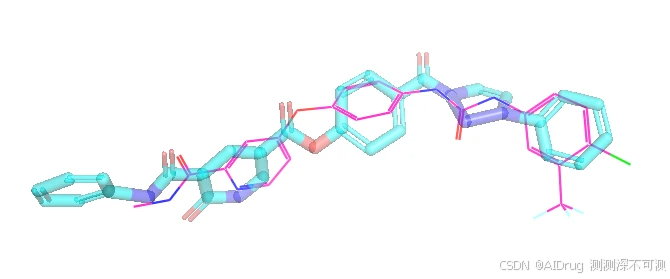
令人惊喜的是,当生成分子与参考分子进行叠合,可以发现,药效团原子(N 和 O原子)的位置基本没有发生变化或仅有少量的位移,如下:
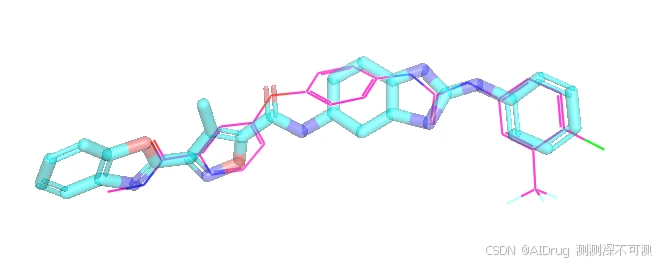
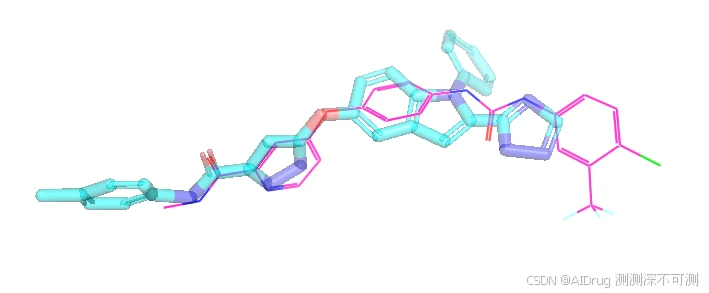
四、总结
本研究提出了 MolSnapper,是一种用于为扩散模型提供3D药效团约束的创新方法,提供了一种在分子生成中实现受控设计流程的工具。MolSnapper 使得利用先验知识来改进分子设计变得更加容易。
(1) MolSnapper 的表现优于 SILVR 和 DiffSBDD 等竞争方法,生成通过 PoseBusters 检测的分子数量最多可提高3倍。此外,在形状和颜色与参考配体的相似性方面,MolSnapper 提升高达20%,从而在初始命中检索上提高了 30% 。
(2) 此研究填补了一个关键空白,使得在大规模分子数据集上训练的模型能够用于结合位点的条件化生成。MolSnapper 通过集成 3D 结构信息和专家知识,实现了一个受控且高效的配体生成过程。
从上述结果来看,MolSnapper 生成的分子在构象上更合理,更像一个真实的构象,没有出现很奇怪和扭曲的化学键。从 qvina 上看,生成的分子打分也比较好,优于或者接近之前介绍的 PIDiff,IPDiff 等模型。因此,整体来看,MolSnapper 是一个更接近真实药物设计需求的分子生成方法。




















 3447
3447

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











