






本篇博客主要是我参与学习EMBL-EBI关于AlphaFold培训课程的一些笔记,
官网相关教程信息,参考:https://www.ebi.ac.uk/training/online/courses/alphafold/
这个教程适合什么样的人去阅读学习:

以及你期望能够从中学到什么:

一,预备知识
1,术语表:
https://www.ebi.ac.uk/training/online/courses/alphafold/glossary-of-terms/
2,第一性原理:Anfinsen’s dogma
安芬森法则:蛋白质结构的“序列决定论”。
其关键价值在于为蛋白质结构预测提供了理论基础 —— 既然序列是结构的唯一 “密码”,理论上就可通过序列反推结构。



3,构象搜索悖论:Levinthal’s paradox
蛋白质链理论上可采取的构象数量极其庞大(如一个含 100 个氨基酸的蛋白,构象数可达10^30以上)。若蛋白质通过 “逐一尝试所有构象” 的方式寻找天然结构,所需时间将远超宇宙寿命(约1.38×10^10年),这与现实中蛋白质能在毫秒至分钟内完成折叠的事实形成矛盾,即 “莱文塔尔悖论” 。
其本质是揭示了 “全遍历构象” 的不可能性,倒逼科学界思考蛋白质折叠的高效路径(而非随机搜索,说到随机搜索,其实我们都会直接想到进化算法、遗传算法之类的实现)。



悖论在于探索构象需要的时间非常长,也就是folding process是一个非常hard的问题,但是也许我们可以避开folding process直接预测folding result。
4,结果和过程并不矛盾
为何安芬森法则与莱文塔尔悖论并不矛盾?
二者看似冲突(前者说“序列决定结构”,后者说“构象太多无法搜索”),实则针对蛋白质折叠的不同维度,核心矛盾可通过“目标与路径的分离”化解:
- 关注对象不同:安芬森法则聚焦“结果”——蛋白质的最终天然结构由序列决定,不涉及“如何达到该结构”的过程;莱文塔尔悖论聚焦“过程”——若通过随机搜索构象的方式达到天然结构,会面临时间困境。二者分别回答“结构由什么决定”和“如何高效形成结构”,不存在逻辑冲突 。
- AI预测的实现其实在实践上进一步验证了“无矛盾性”:现代AI(如AlphaFold)通过学习大量已知的“序列-结构”对应关系,可直接预测蛋白质的最终三维结构,无需模拟折叠过程——这正是安芬森法则的实践体现(序列→结构),同时也避开了莱文塔尔悖论的“过程困境”(无需遍历构象) 。
- 理论与现实的统一:安芬森法则为结构预测提供了“可行性”(序列含结构信息),莱文塔尔悖论则指出“直接遍历构象不可行”,二者共同推动了对“高效折叠路径”的研究(如能量漏斗模型、分子伴侣机制),最终形成“序列决定结构,高效路径实现结构”的完整逻辑 。
综上,安芬森法则与莱文塔尔悖论并非对立关系,而是从“结果”与“过程”两个维度共同揭示蛋白质折叠的规律:前者确立了“序列→结构”的理论基础,后者则倒逼科学界探索高效折叠路径,二者共同推动了蛋白质结构预测(尤其是AI预测)的发展。
5,3个数字:2020,CASP14,Alphafold2
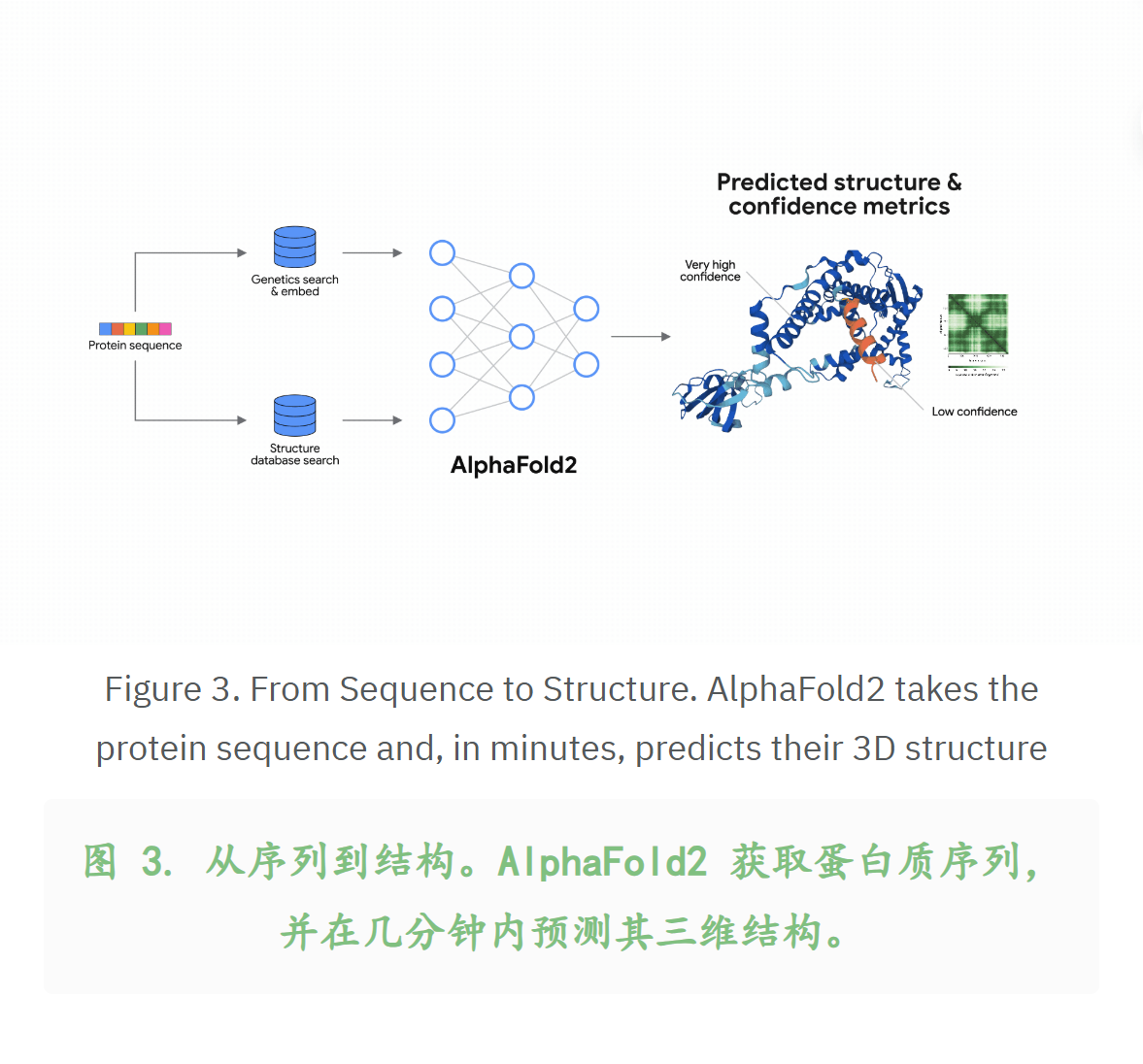

6,如何访问
alphafold3网页服务器:https://alphafoldserver.com/
colabfold(让每一个实验室都能够做结构预测)https://colab.research.google.com/github/sokrypton/ColabFold/blob/main/AlphaFold2.ipynb#scrollTo=kOblAo-xetgx
现成的alphafold数据库:https://alphafold.ebi.ac.uk/
7,机器学习的角度:理解Alphafold能做什么不能做什么?







下面这个例子其实就是动态/生理建模,通过RMSD误差的评估其实我们可以看到差异所在:
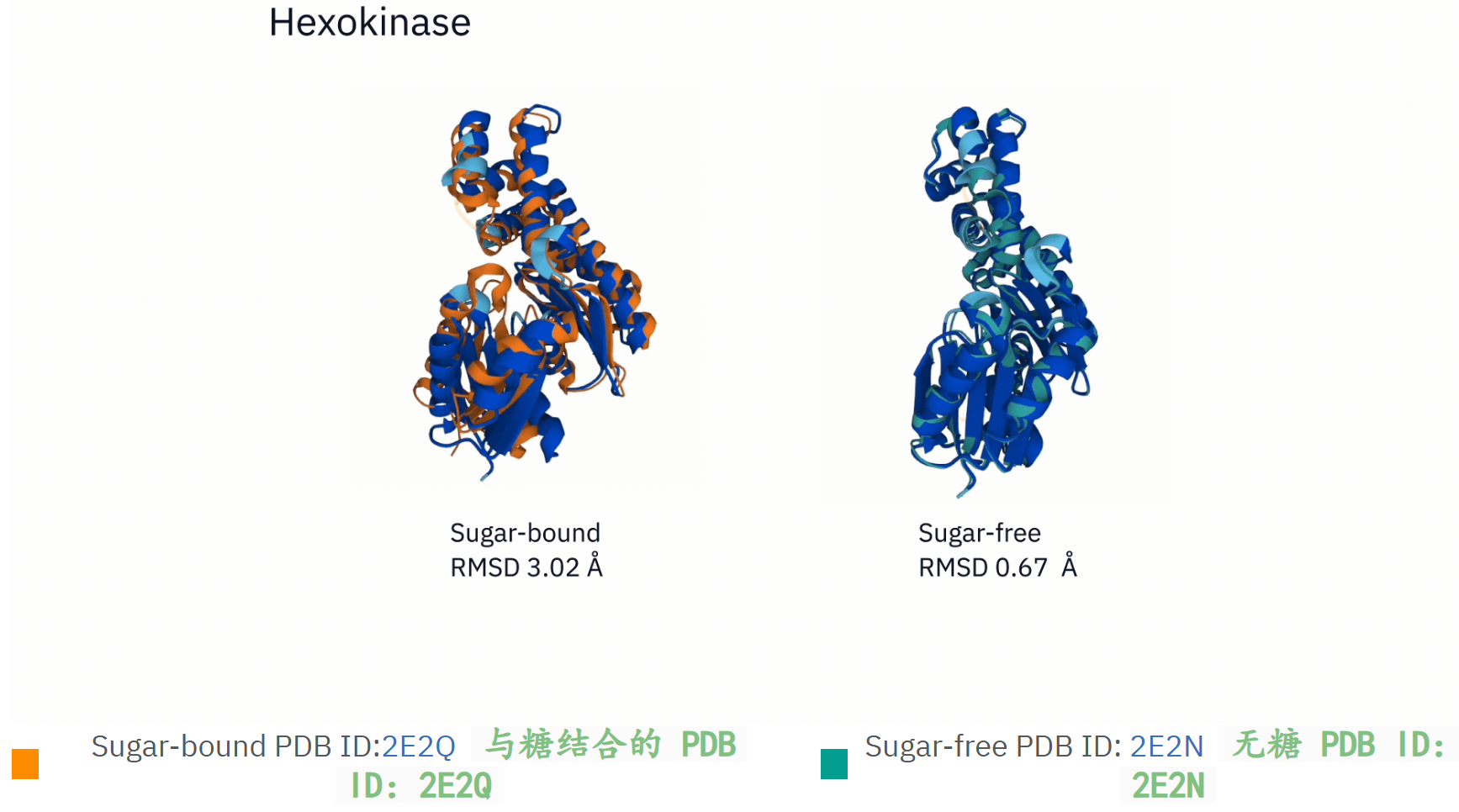




二,验证与影响
这一节开始单独与预备知识抽离出来,是因为这一节开始就是实践应用的人应该重点考虑的相关因素了。

1,AlphaFold2 的蛋白质结构预测是如何得到验证的?


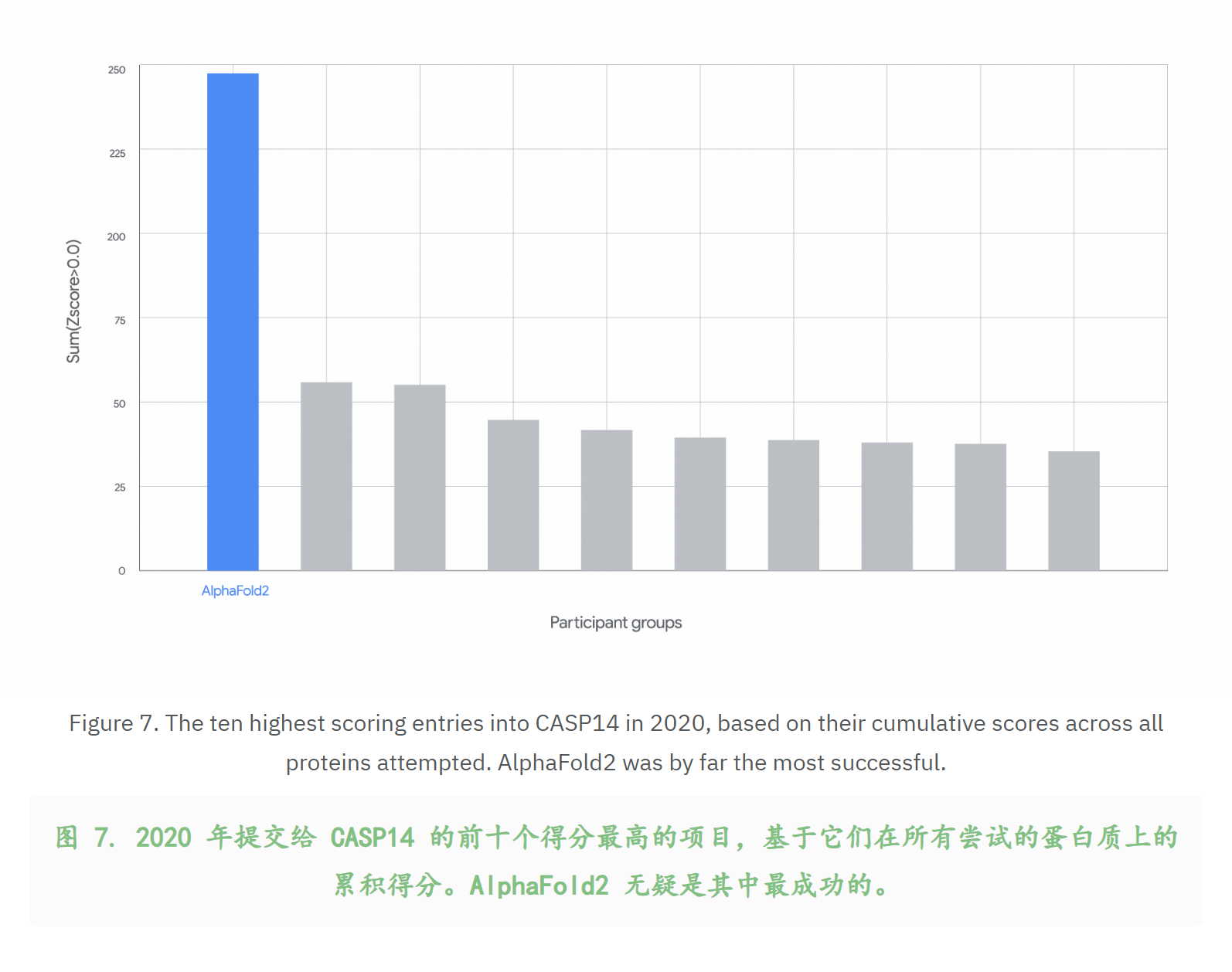

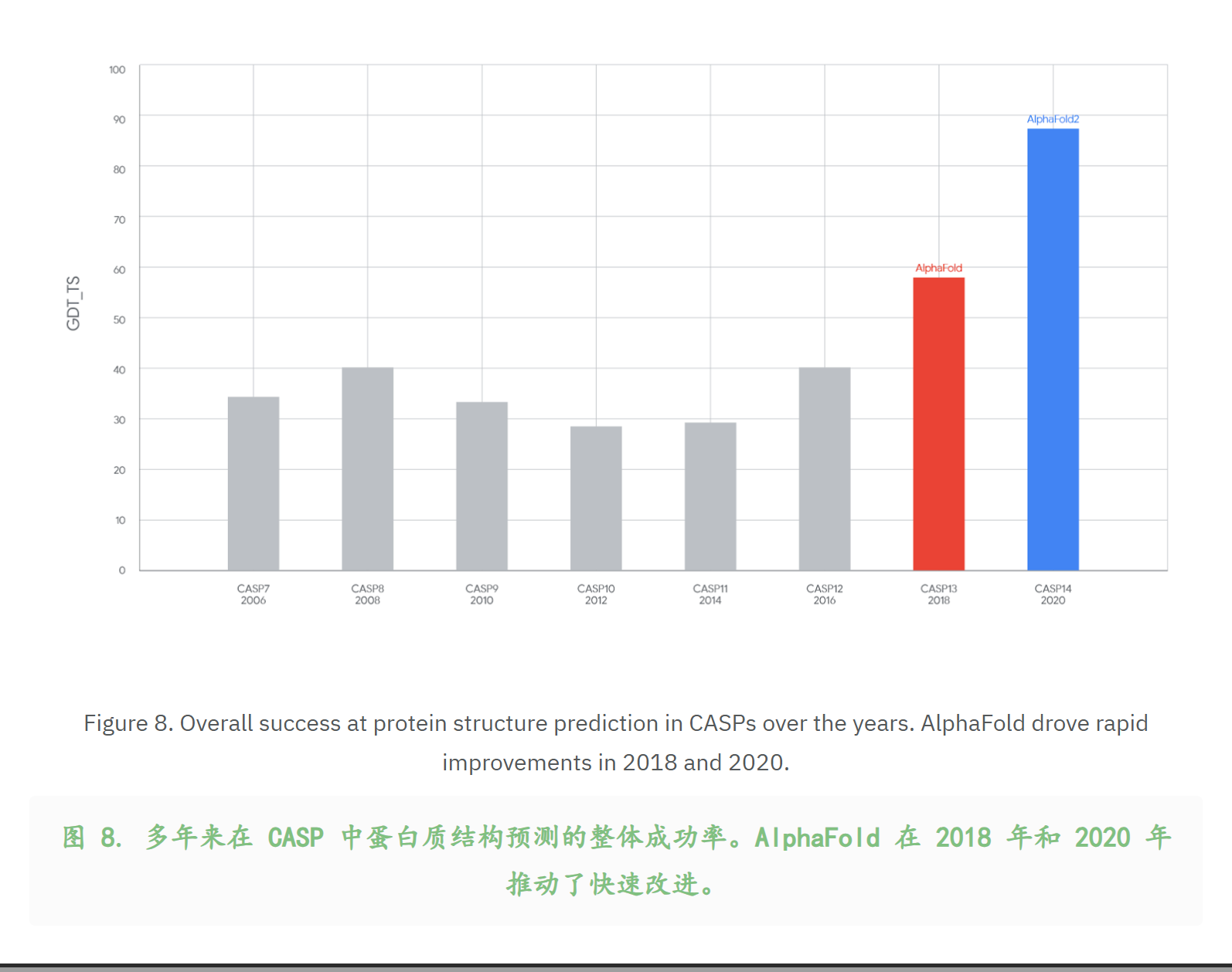
CASP15 alphafold系并没有参加,但是当时alphafold已经开源了。

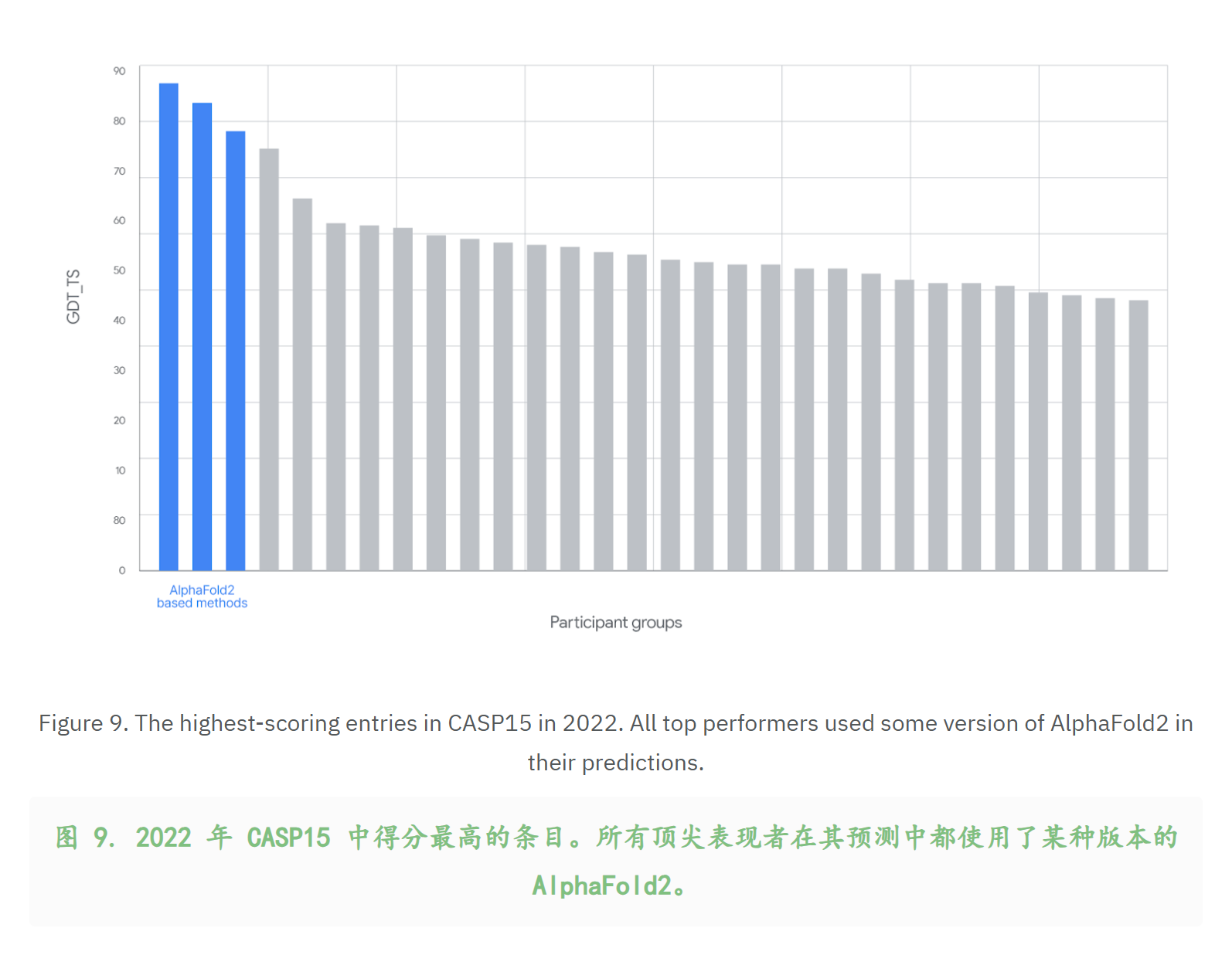
结构生物学的后续证据


其中溶液和结晶是从机器学习的角度来看的,毕竟如果假设这是两个蛋白质构象的数据分布,那么只在结晶数据PDB上训练的model是不是会有分布bias偏差?
其实这一点一直被人argue,也基本上是个搞机器学习的都会问这个傻二愣问题,但是这个现象其实早就有研究解释了。(所以,下一次,如果你科普alphafold模型,或者是做类似model的汇报,再有人问你训练数据是结晶巴拉巴拉,会不会有bias的时候,你直接用文献报道回怼回去,我见到过不少做湿实验的老师问过这个问题,总感觉还活在上一个世纪一样)。


2,AlphaFold 2 的结构预测有多准确?



前面是讲RMSD指标用来评估测得准不准,后面就是举几个例子说明哪些是通常测不准的:
侧链、IDR无序区域、结构域之间的柔性连接体linker、跨膜蛋白



3,科学家如何使用 AlphaFold 2?或者说,科研使用AlphaFold2的正确姿势是怎么样的?
Structural biology 结构生物学干湿并行



使用 AlphaFold2 结构预测指导分子和细胞生物学


蛋白质工程与设计


三,输入与输出

重点是理解下面这些指标:


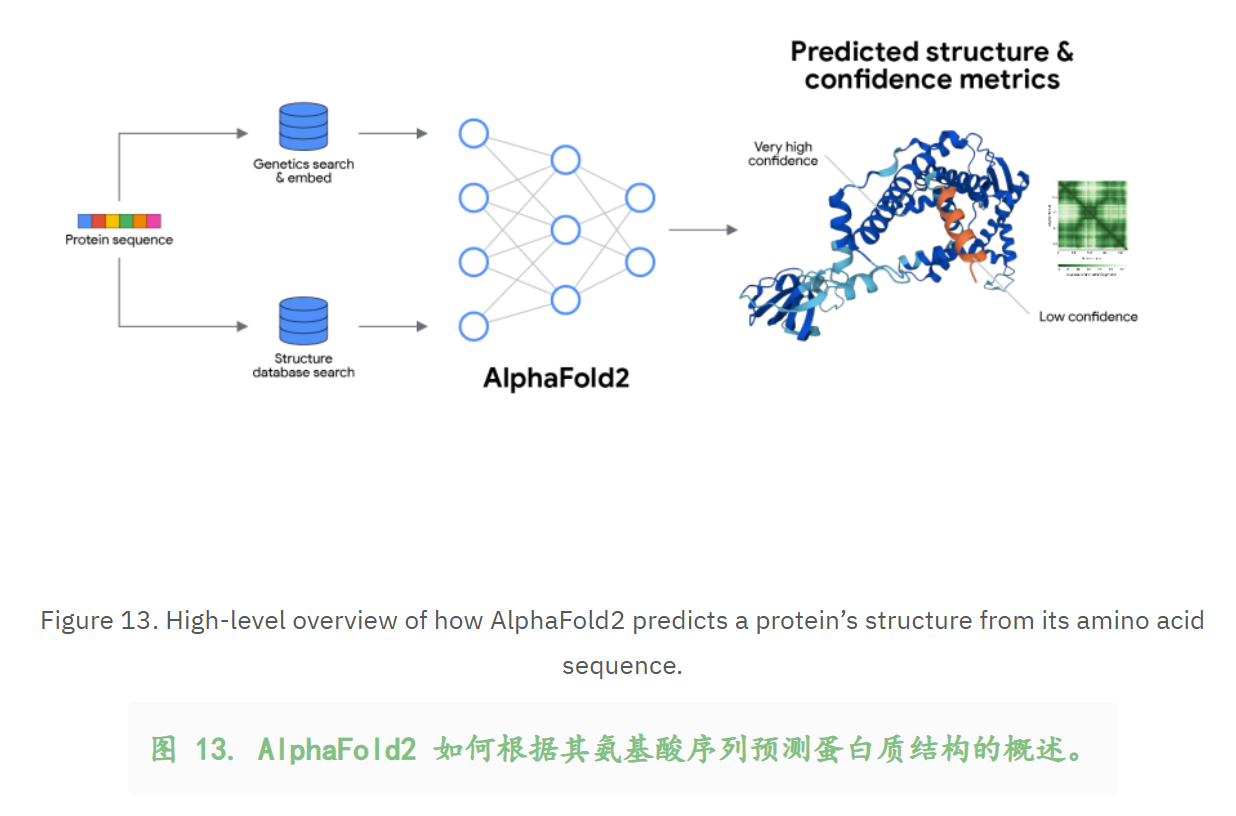
1,多序列比对MSA的作用


2,成对表示的作用

3,如何获取最终的结构?

4,使用置信度分数评估 AlphaFold2 预测的结构

部分可以参考我之前的博客:https://blog.youkuaiyun.com/weixin_62528784/article/details/144540998?spm=1001.2014.3001.5502
但是这篇博客基本上是一些二手信息解读,其实并不是很准确,所以还是以本篇博客为主,是一手信息。
pLDDT:理解局部置信度

局部距离差异测试,参考:https://academic.oup.com/bioinformatics/article/29/21/2722/195896?login=true
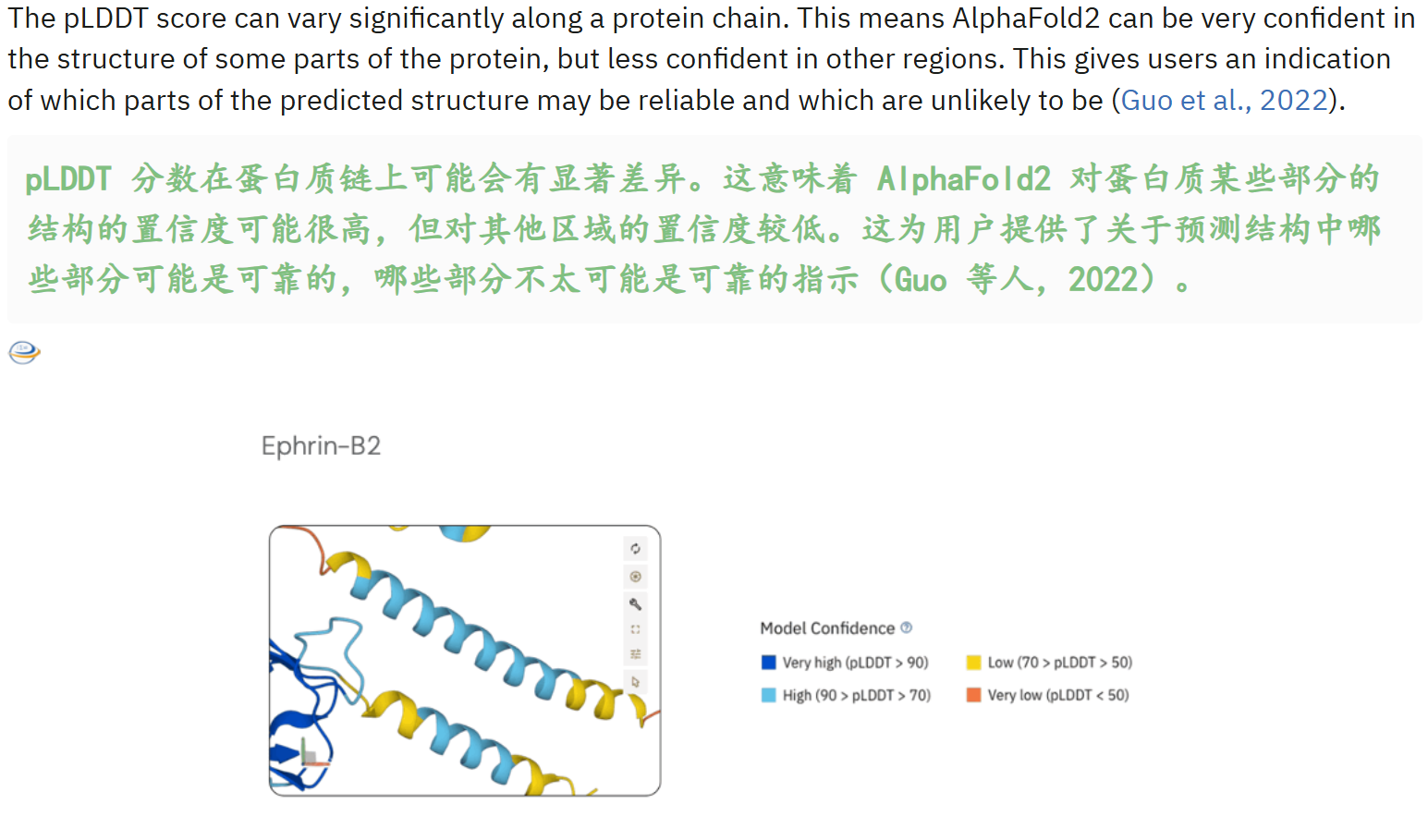
现在我们获取的网页服务器预测结果,一般对于pLDDT的测试效果如下:
基本上看青色、蓝色就可以了。


以及还是老问题,对于IDR也就是内在无序区域的序列部分:

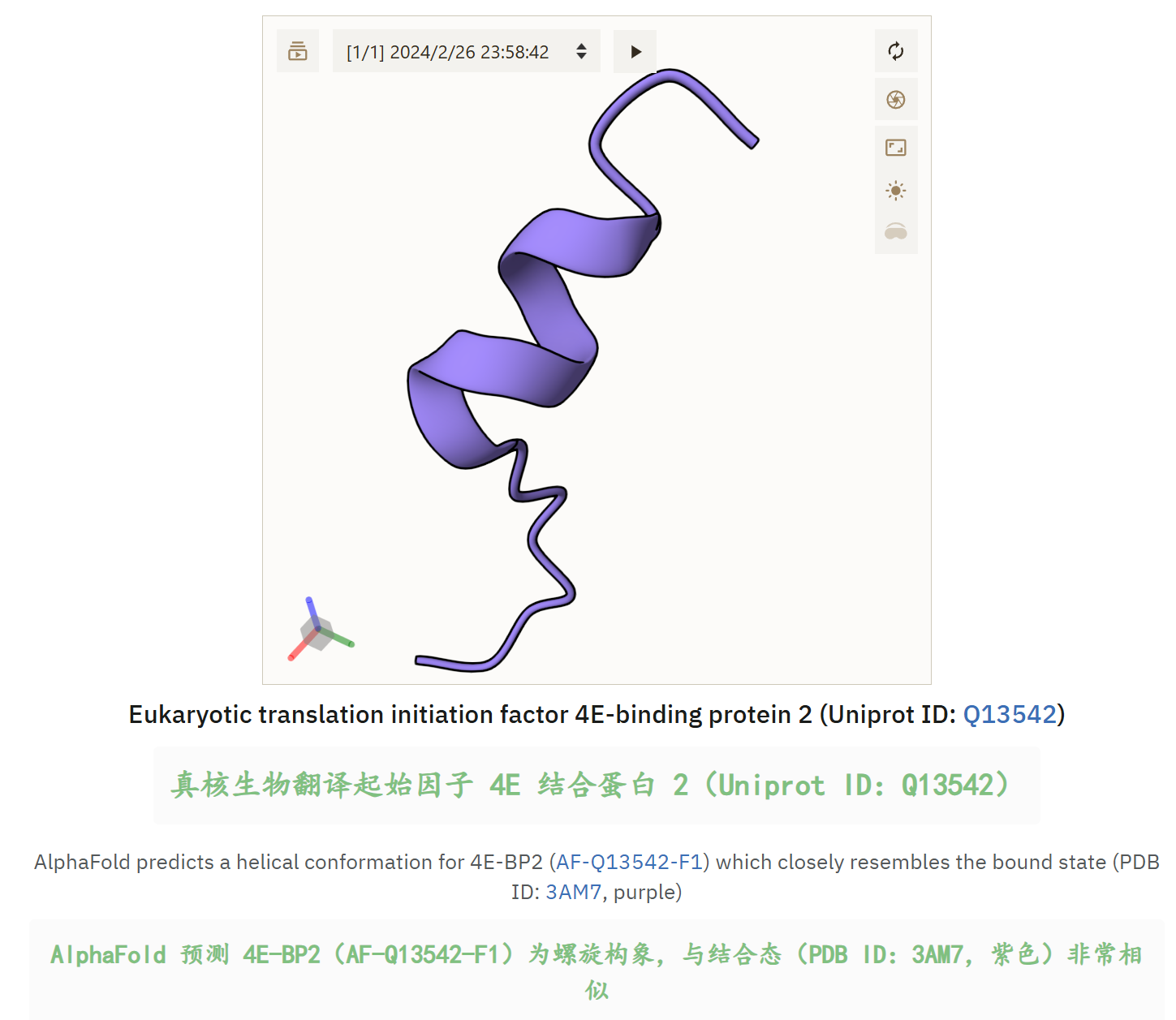
最后还是要说明,所谓高置信度还是局限在局部指标下,比如说结构域局部置信度高,那么涉及到局部的好说,但是结构域与另外一个高置信度结构域之间的相对位置,这个指标其实是跨结构域的,也就是非局部指标,
非局部scale,不好拿过来评测。

几个预测实例:



PAE:AlphaFold2 预测的全局置信度度量
前面说了,pLDDT是局部置信度指标,只能够拿来分析以及查看局部的结构是否预测可靠,涉及到跨局部的指标、特征以及其他结论,都不能够直接推导。
而PAE,就是在全局层面进行描述的一个置信度指标,

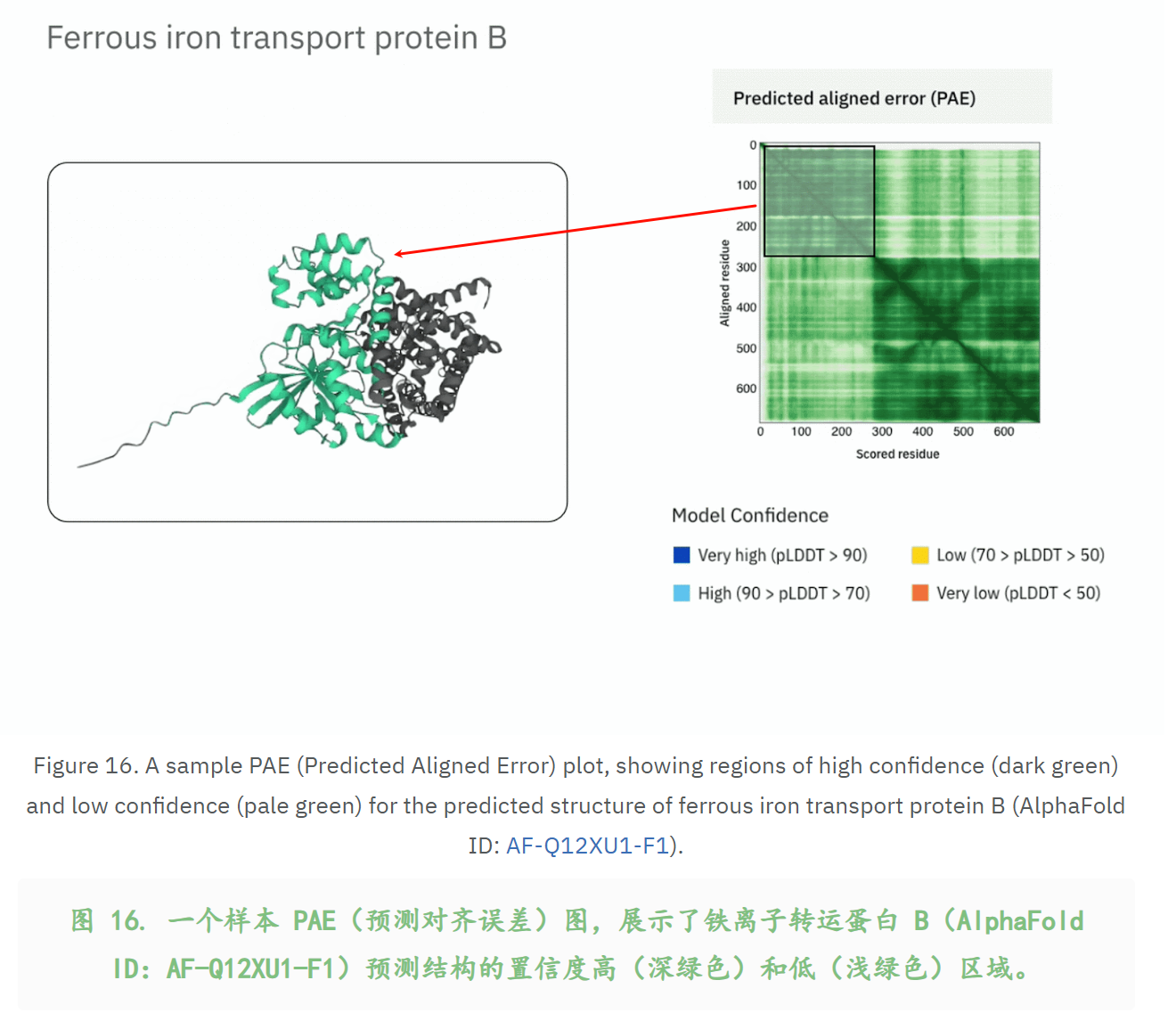
比如说下面结构预测的PAE图,就有两个深绿色的矩形区域,以及其他两个浅绿色的矩形区域;
这个图怎么来看:在y轴处画一条横线,也就是预测结构和实际结构在这条横线上的y残基处对齐重叠,这个时候x轴残基处两个结构之间(预测和实际)会有一个距离,也就是有一个差异值,这个差异值有一个分布,取均值就是PAE值,我们在(x,y)处将这个PAE值映射/压缩成一个颜色色深的数值,比如说是浅绿色还是深绿色。
所以,从定义我们其实可以看到,这个指标是用来描述一个残基对(x,y)的相对距离的置信度指标,
这么来讲吧,如果我预测的结构中(1,727)也就是第1位和第727位的残基,如果我在727位置上将预测的结构和真实的结构对齐重叠了,如果这个时候第1位的残基也是重叠的,也就是PAE的预期值是0,那不就正好说明1、727这两个残基之间的位置正好被我的model预测准确了吗,因为我建模中这两个残基的位置,也正好都(注意是都,也就是两个)被真实结构所对齐重叠了。
所以说(x,y)处PAE值越低,也就是置信度越高,说明我的模型在估计第1和第727这两个残基的相对位置是比较准确的,是和真实结构相差不大的。
那么我们可以外推,对于一个置信度比较高的矩形区域,也就是下面这种深绿色的区域,这意味着什么?
首先,一个深绿色的点(x,y),表明model预测x、y之间相对位置比较准确;
那个一堆点呢,表明什么?
比如说(x1:xn)x (y1:yn),就表明x1-xn这个区域和y1到yn这个区域,这两个区域之间任意一对点,也就是任意一个残基组合,预测都是比较准的,
也就是说对于x1-xn、y1-yn这两个区域之间残基群体,这个群体的内部空间组织,我是有把握的,它们怎么组织、空间距离如何,我都是有把握、预测的都比较准确的。
我们再反推一下,什么样的区域内(就什么样的一个残基群体),我们是有把握预测一种内组织/自组织/空间群体结构稳健的规律的?
其实就是结构域,因为一个结构域比较保守,所以一个结构域内每个残基怎么放置、怎么旋转,相对位置如何如何其实都是比较固定的,所以对于结构域,是能够一大片残基群体(两两组合)之间都能够预测把握的比较准的。
所以,对于一个一个的高置信度区域,也就是一个一个的深绿色区域,我们可以简单理解为是一个相对自组织完好/空间组织相对固定/可能比较保守的一个空间组织单元,或者简单理解为一个结构域。
当然,我上面的这些看法只是通俗地解释一下,仅供参考。
(另外注意一下PAE这个图不是对称的!!!看看定义就能够想明白了)
此处再次强调,PAE这个指标非常重要!pLDDT是看局部置信度的,我们只看在局部用这个指标,那么全局我们必须得用上PAE的信息!

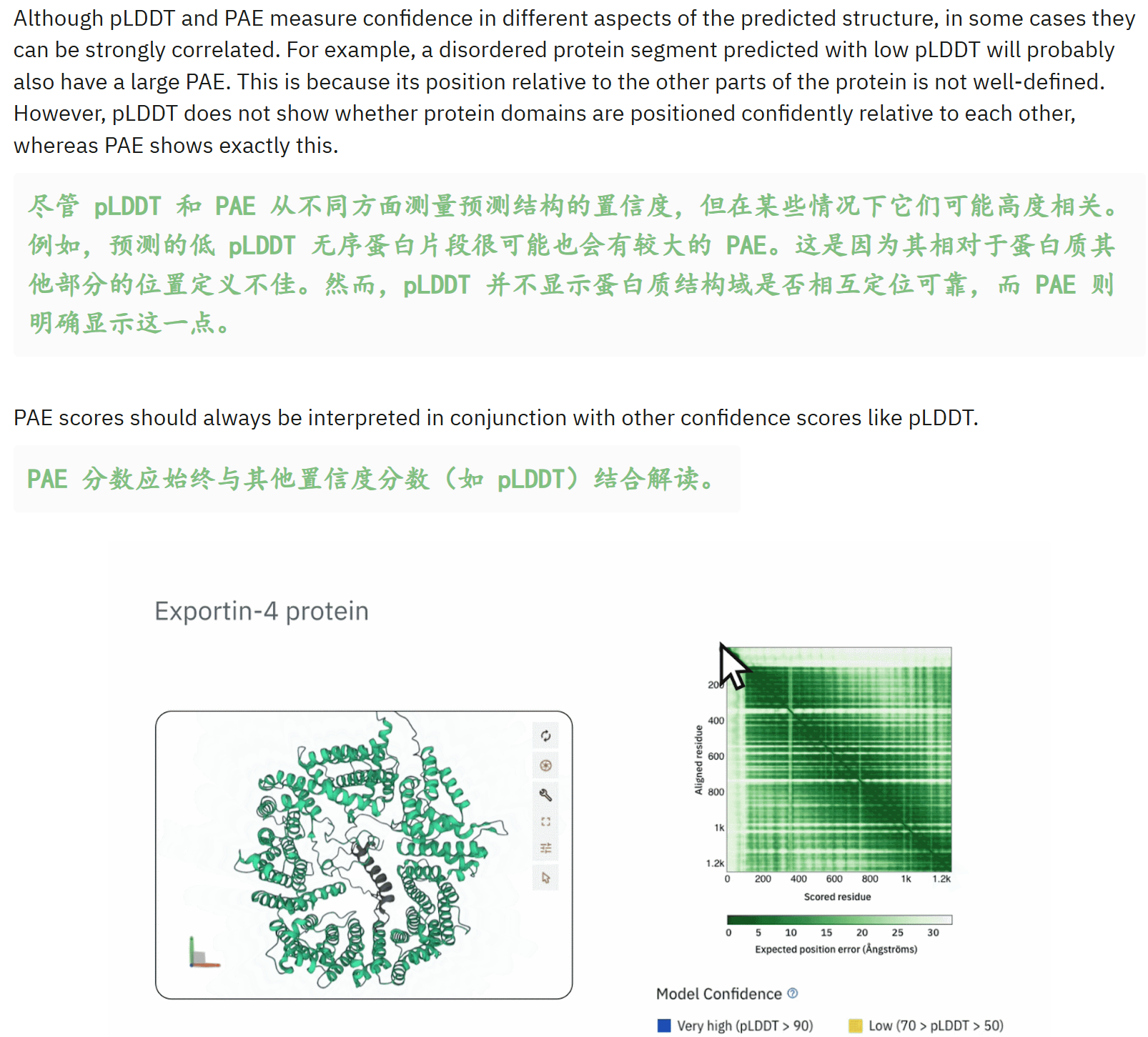
官网给了一个非常简洁却有意思的例子:
下面是一个蛋白质的预测结构,我深绿色标注出来的就是两个结构域,因为结构域内部组织比较保守,所以PAE会比较高(这一点我前面通俗解释过了)。
一般常人看到这个结构,脑回路一般是这样的:
哇,pLDDT好高的两个结构域,肯定可信(PAE分数也没问题,局部一团绿)!
哇,居然距离还离得这么近,一定说明这两个结构域是互作的!
两个哇叠加在一起,简直是喜上加喜,我立马有了一个idea,就叫做xx蛋白中两个结构域的互作!
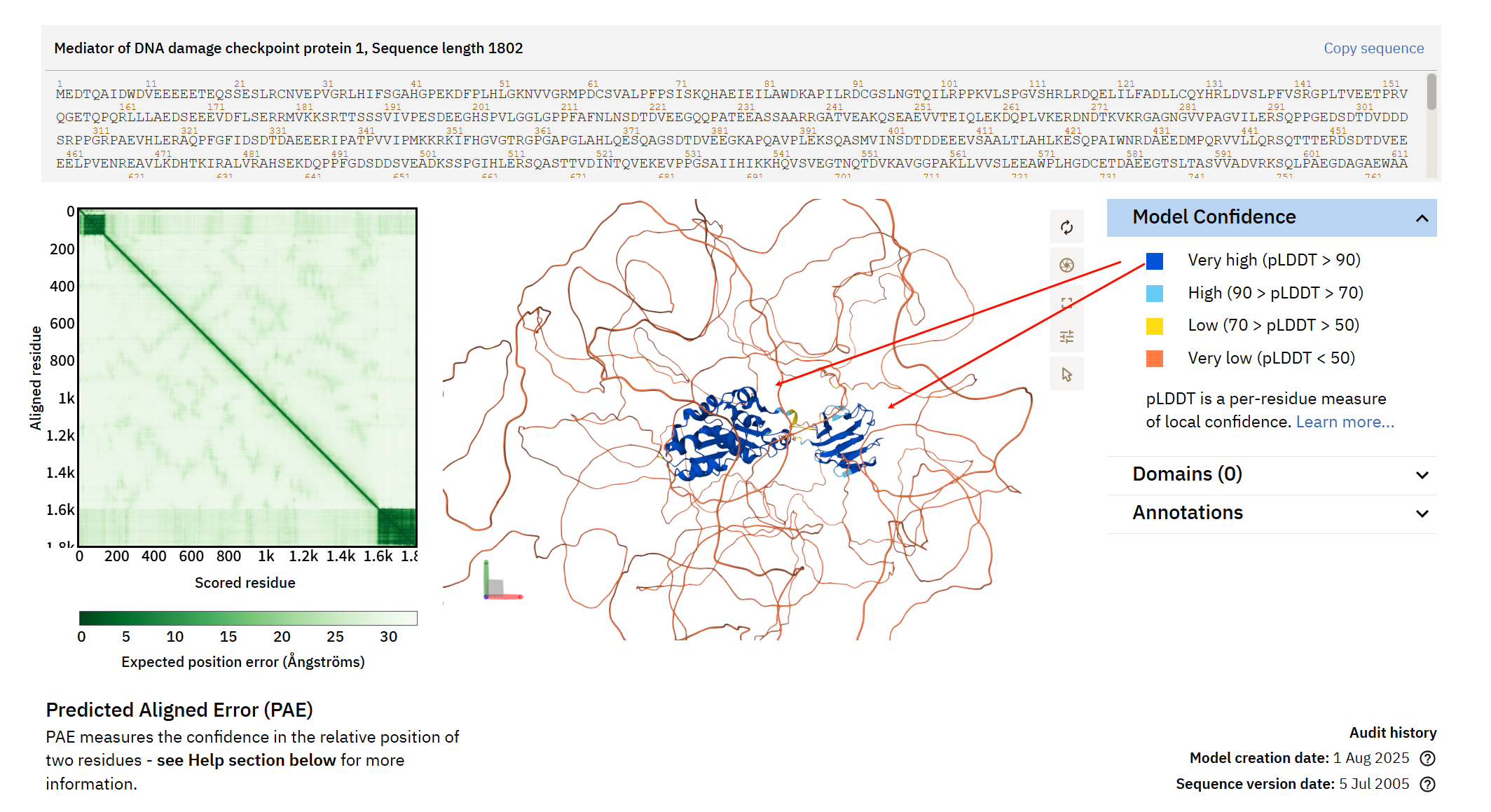
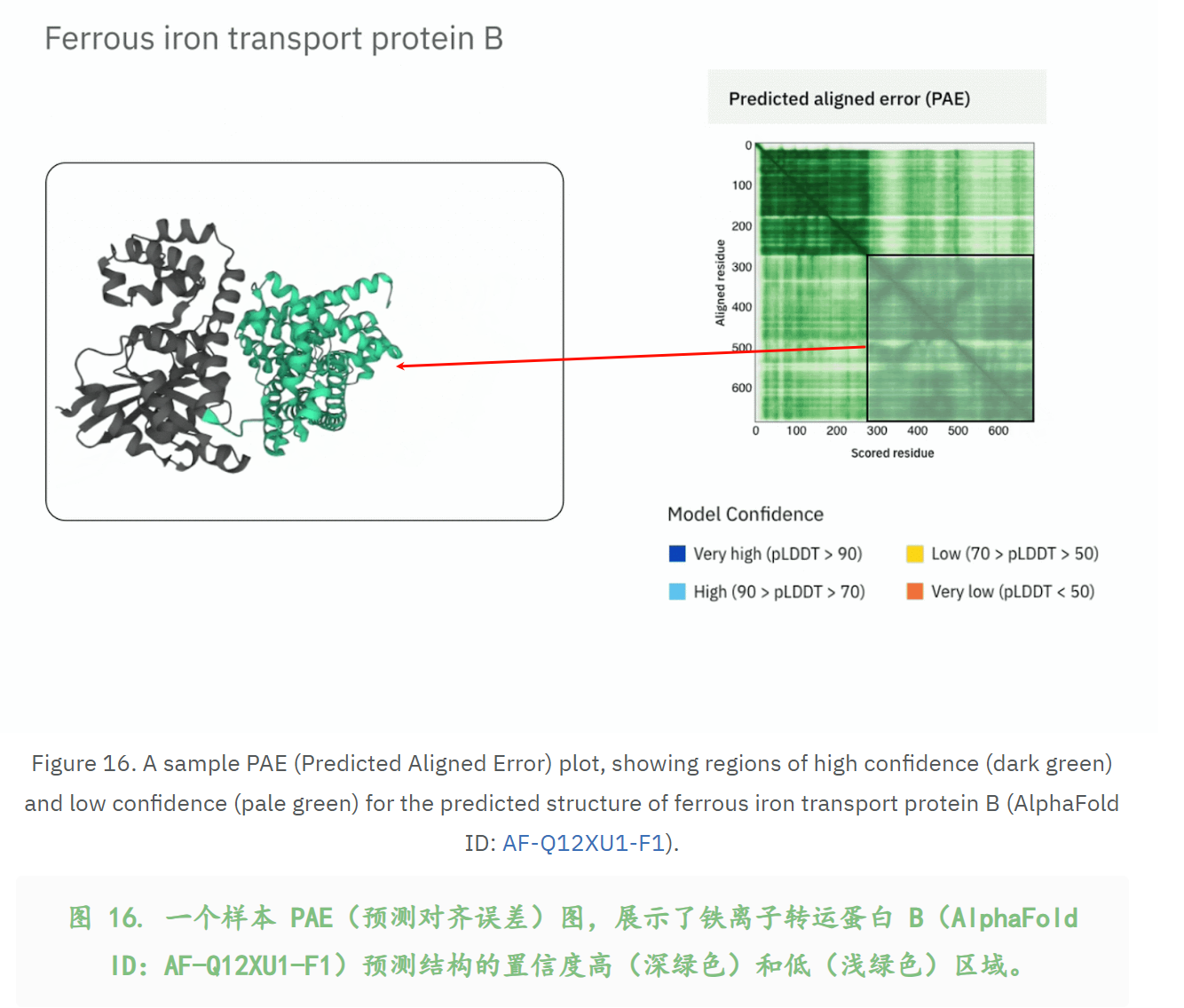
然而,结果是错,而且是大错特错!
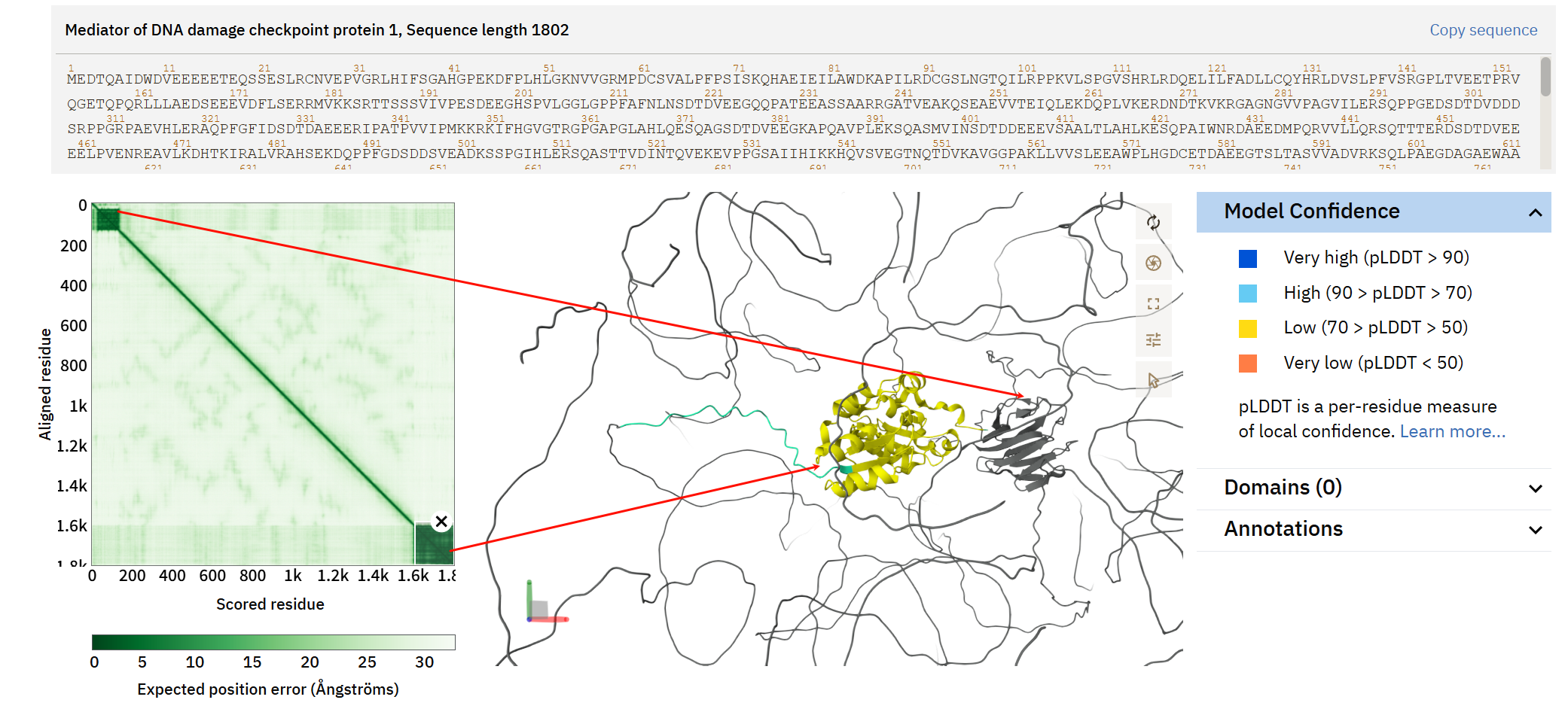
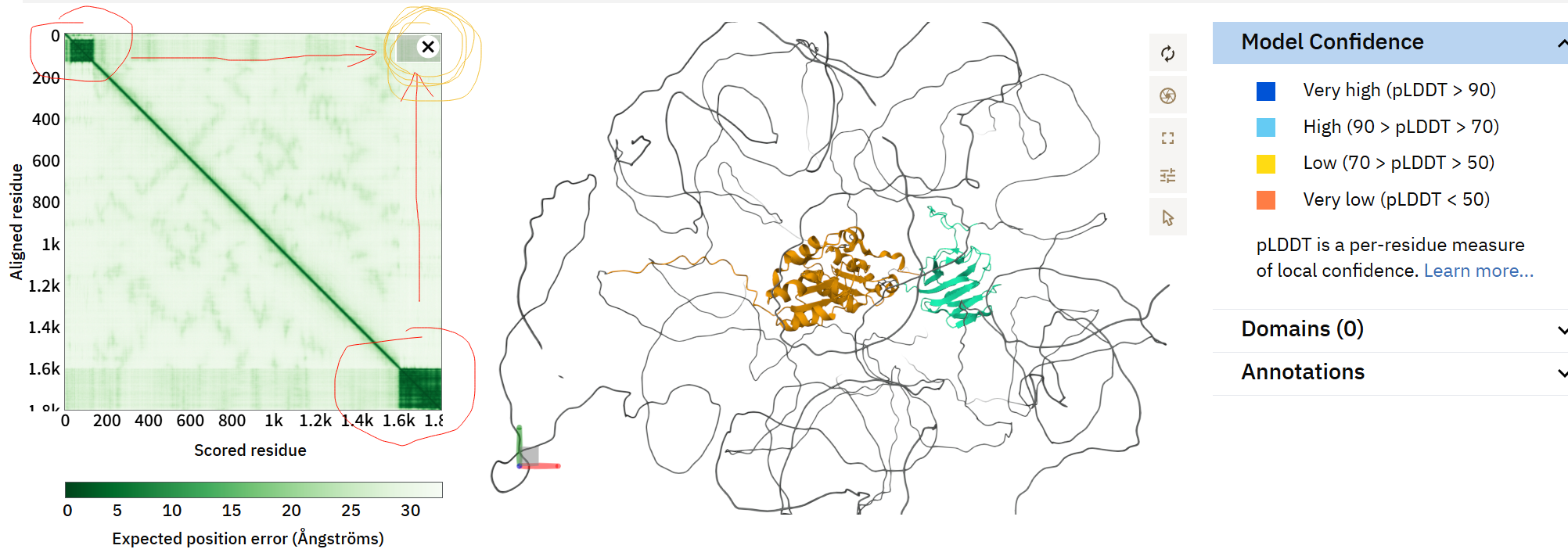
为什么,到底很简单,看看这两个结构域所代表的区域之间的PAE分数,
想象一下,把第一个结构域抽象成region1,第二个是region2,我们来看一下region1 x region2这一块区域,
就是下图我圈出来的这个橙色圆圈,我们可以看到(region1,region2)这个区域的PAE分数非常低,说明对于region1和region2之间的相对距离,我们的预测结果是相当没有把握,哦豁,我们的假设泡汤了!
(总而言之,将区域抽象成点,就是对称轴上要看,区域交叉我们也要关注)


下面是几个官网的示例解读:
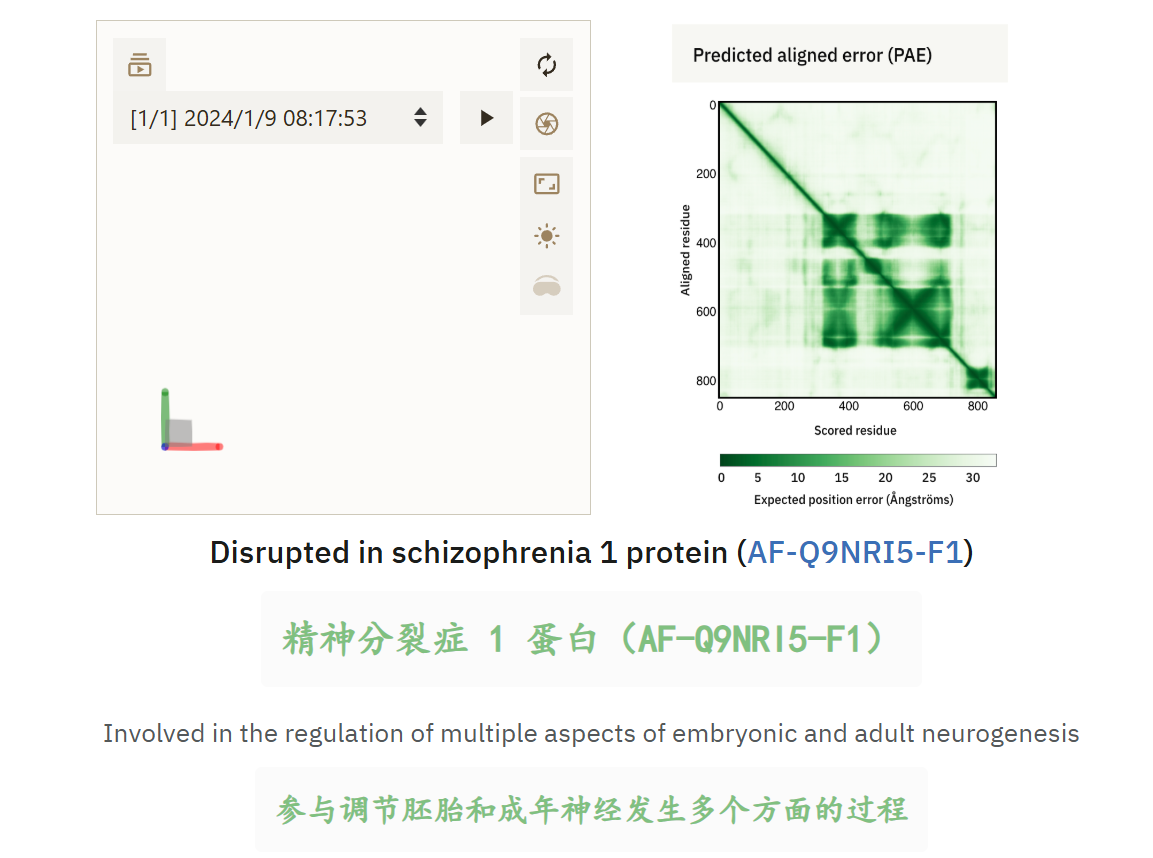

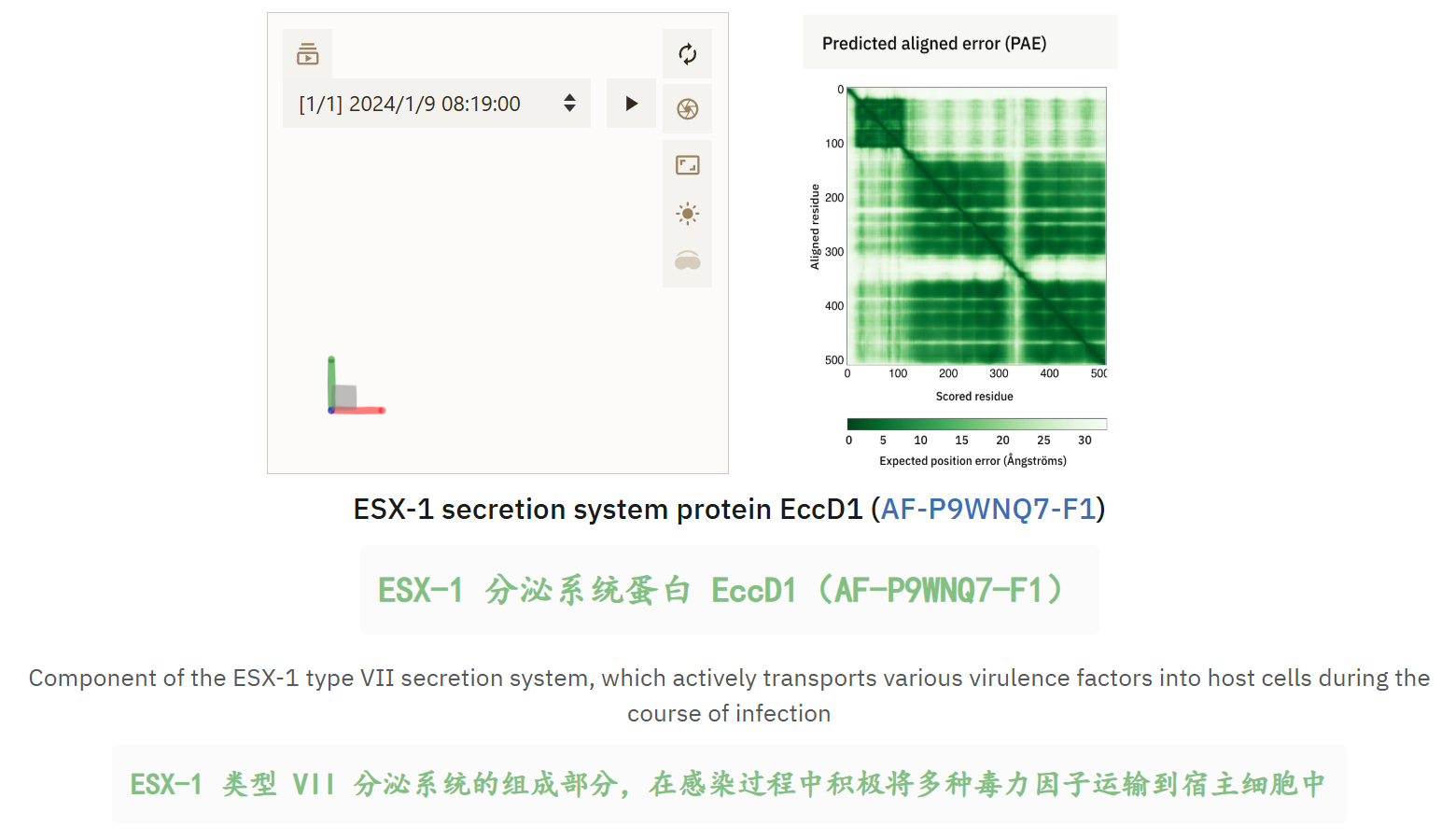

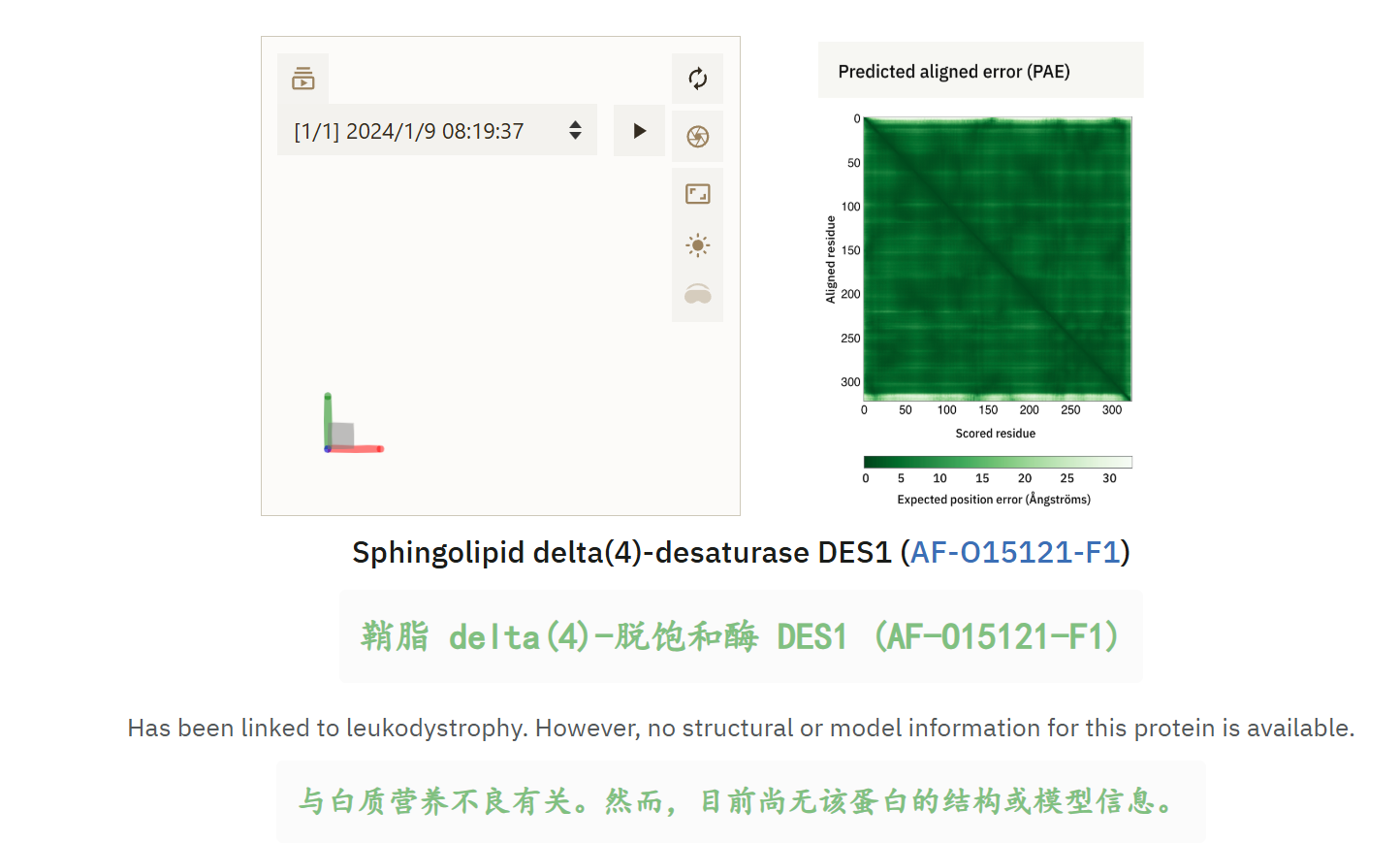

总而言之,需要整合pLDDT和PAE两个指标来看。
pLDDT是看局部结构可不可靠,也就是order/disorder,局部一个东西是不是真的?
PAE是看局部结构之间的相关关系、相关定位准不准,两个真的东西之间的摆放相对关系是不是真的?
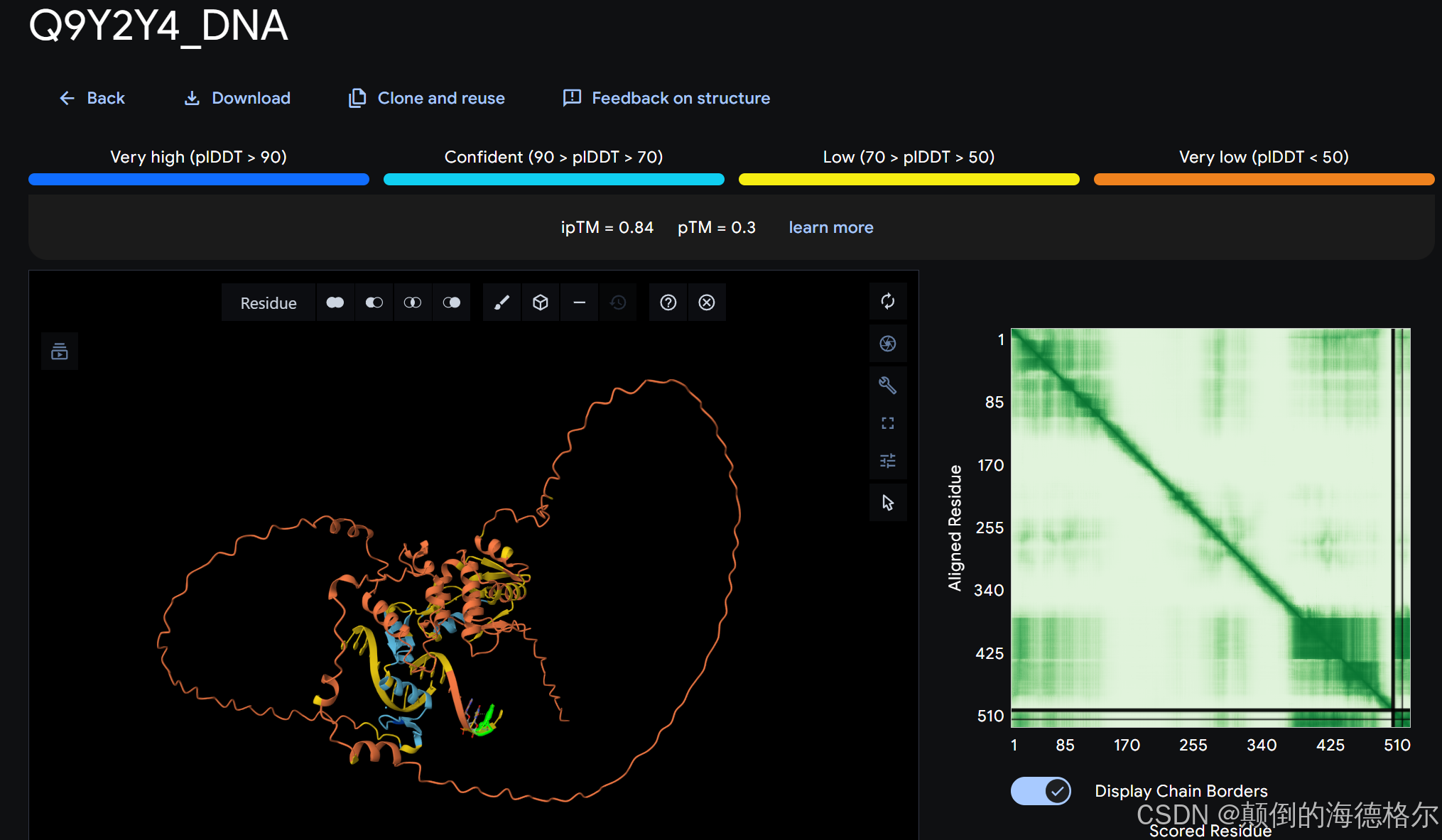
AlphaFold-Multimer 中的置信度分数
主要是复合物预测这方面提出了两个新的置信度指标,
pTM是指复合物整体结构预测的准确性,然后以0.5为一般的判断标准;


ipTM也是一个结构相对位置的指标,而且官方明确说明ipTM这个指标可能比pTM更有用。

比如说我手头上有这么一个预测结构:
一般给出来的4个指标都全了,我个人是习惯先看pTM,先看看整体可不可靠(iPTM我一般不怎么看,暂时也没想到怎么看),如果pTM还行,我需要深入每一个结构,那我再看pLDDT也就是结构中哪些区域可靠,如果可靠的话,我再看这些pLDDT可靠的结构域之间的PAE是否可靠? 至于复合物之间,也就是大单元之间的iPTM,只有一个数值,暂时没想到放在什么顺序看比较好。

















 517
517

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









