







 RNA-seq是一种基于二代测序技术研究转录组的工具,用于定性和定量分析基因表达。该技术涉及数据质检与控制、接头去除、基因映射、表达量量化等多个步骤,常用工具包括FastQC、Cutadapt、STAR等。数据分析包括基因表达差异检测和标准化,如FPKM和TPM的计算,广泛应用于生物学和医学研究。
RNA-seq是一种基于二代测序技术研究转录组的工具,用于定性和定量分析基因表达。该技术涉及数据质检与控制、接头去除、基因映射、表达量量化等多个步骤,常用工具包括FastQC、Cutadapt、STAR等。数据分析包括基因表达差异检测和标准化,如FPKM和TPM的计算,广泛应用于生物学和医学研究。
RNA-seq介绍
RNA-seq(RNA sequencing)是一种基于二代测序技术(Next-Generation Sequencing, NGS)研究转录组学的方法,能够快速获取给定时刻的某一基因组中RNA的种类和数量,从而实现定性与定量分析。
随着二代测序技术的发展,RNA-seq的应用场景也不断拓展,在生物学和临床医学等领域都有广泛应用。如今,RNA-seq在诊断、预后和治疗各种疾病(包括癌症和传染病)的应用中具有广阔前景。
RNA-seq数据分析的基础是下机数据的质量检查与控制(简称“质检、质控”),即对测序质量总体上有所了解,并进行质量控制,保留满足后续分析的高质量数据。不同的生物信息学工具(如FastQC)能够对测序数据整体及评分,如Phred质量评分、读长长度分布、GC含量、接头含量、重复读长等。在需要去除接头时,可以使用Cutadapt,Fastp和Trimmomatic等工具。
下一步,使用splice-aware算法将原始数据reads映射到人类参考基因组,例如STAR、TopHat2或HISAT2。此时,必须根据研究类型和表型调整一些重要的变量,如基因组的参考版本、参考基因组的注释文件等。为了丰富参考基因组注释,可以通过融合多个数据库得到较为全面的背景数据集。
在将读数映射到基因组后,有一些技术和生物学偏好会影响灵敏度阈值。比对结果中3’末端基因的偏好性可能表明RNA的降解,或者表明数据来自3’测定(例如,寡聚T起始的3’RNA-seq),可以使用RSeQC等工具来评估BAM文件中基因的覆盖率。
RNA-seq最广泛的应用就是用来评估基因和转录本的表达,这一应用主要是基于比对到转录组区间内的读长的数量。例如HTSeq,FeatureCounts和Salmon,这些工具可以量化特定基因特征中的映射读数。其中的一些偏差,如基因长度或GC含量可能会影响量化的过程,并对差异表达分析(DEA)产生负面影响。目前已经有几种方法,能够减少这些偏差。包括基于基因长度和文库大小(每次复制的读取总数)将读长计数归一化。常用的方法包括对单端读长以RPKM(每千个碱基的转录每百万映射读取的读长)为单位,双端读长以FPKM(每千个碱基的转录每百万映射读取的片段)作为单位,和以TPM(每百万映射读取的读长)作为单位。
基因表达水平定量之后的分析可使用相应的软件包来实现。差异表达分析可选择NOIseq识别RNA-seq数据中的误差来源,并使用相应的方法来标准化这些误差,同时也可以使用COMBAT和ARSyN等及进行批次矫正。此外还可使用edgeR、DESeq、baySeq和EBSeq等进行分析。可变剪切分析可以使用BASIS、CuffDiff2、rSeqDiff等进行分析…
许多独立的研究都已经证实,选择不同的方法会对结果有一定影响,而且没有哪一种方法能够适用于所有的数据,所以,在具体分析的时候需要使用多个软件进行相互验证。
数据处理流程
如上文所说,RNA-seq有许多工具可以使用,实际中根据自己的需求选择合适的工具组合。
最近接触的一套数据处理流程如下:
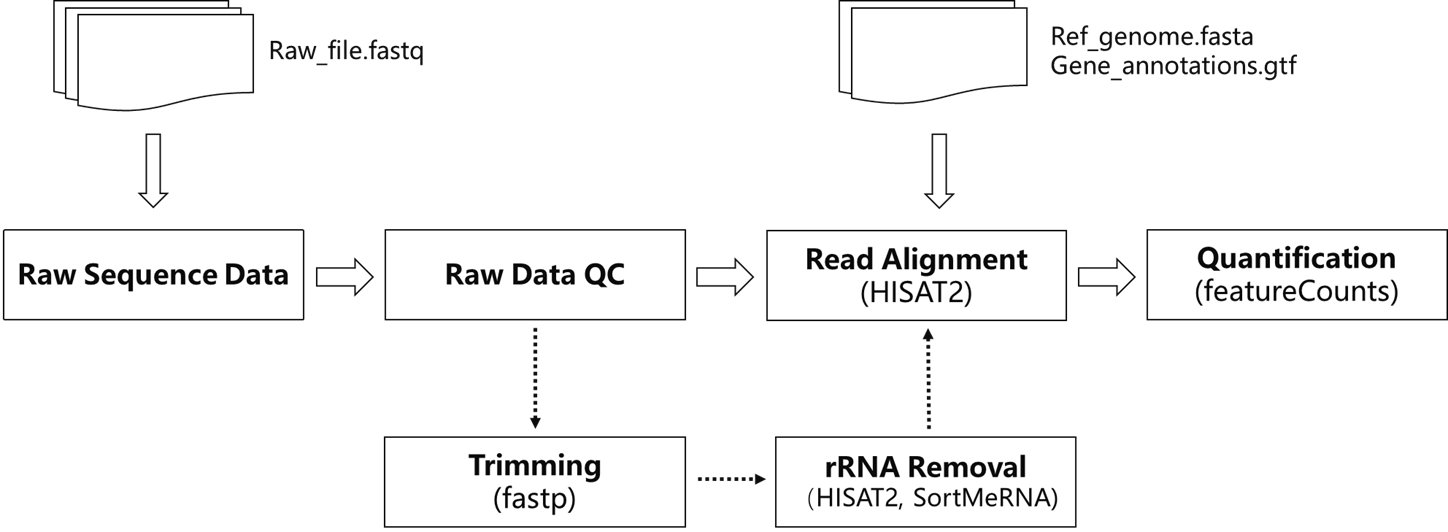
Raw Dara QC
step1 Cut adapter
fastp \\
-i $fq1 -I $fq2 \\
-o $outdir_step1_fastp/N1.r1.fq -O $outdir_step1_fastp/N1.r2.fq \\
-h $outdir_step1_fastp/cutadapter_fastp.html -j $outdir_step1_fastp/cutadapter_fastp.json \\
--adapter_sequence="接头序列"\\
--adapter_sequence_r2="接头序列"\\
--disable_trim_poly_g \\
--disable_quality_filtering \\
--disable_length_filtering \\
--dont_eval_duplication \\
--thread=12 -c \\
--failed_out $outdir_step1_fastp/${id}_cutadapter.failed_out.fq.gz &&
step2 Quality control
fastp \\
-i $outdir_step1_fastp/N1.r1.fq -I $outdir_step1_fastp/N1.r2.fq \\
-o $outdir_step1_fastp/N3.r1.fq -O $outdir_step1_fastp/N3.r2.fq \\
-h $outdir_step1_fastp/quality_filter.fastp.html -j $outdir_step1_fastp/quality_filter_.fastp.json \\
--disable_adapter_trimming \\
--thread=12 \\
--disable_trim_poly_g \\
--qualified_quality_phred=30 \\ ##设置过滤质量值
--unqualified_percent_limit=50 \\
--disable_length_filtering \\
--n_base_limit=10 \\
-p -c \\
--failed_out $outdir_step1_fastp/${id}_quality_failed_out.fq.gz &&
step3 Length control
/fastp \\
-i $outdir_step1_fastp/N3.r1.fq -I $outdir_step1_fastp/N3.r2.fq \\
-o $outdir_step1_fastp/${id}.r1.fq.gz -O $outdir_step1_fastp/${id}.r2.fq.gz \\
-h $outdir_step1_fastp/${id}_lengthfilter.fastp.html -j $outdir_step1_fastp/${id}_lengthfilter.fastp.json \\
--disable_adapter_trimming \\
--thread=12 \\
--disable_trim_poly_g \\
--length_required=17 \\ #设置过滤长度
--disable_quality_filtering \\
--dont_eval_duplication \\
-p -c \\
--failed_out $outdir_step1_fastp/${id}_length_failed_out.fq.gz &&
rm $outdir_step1_fastp/N3.r1.fq
rm $outdir_step1_fastp/N3.r2.fq
step4 rRNA depletion
这里需要自己去制作一个rRNA的索引哈
hisat2 -k 10 -p 12 --no-unal \\
-x rRNA_indedx -S $outdir_step2_rRNA/${id}_accepted_hits.sam \\
--fr --dta --avoid-pseudogene \\
--un-conc-gz $outdir_step2_rRNA/${id}_non_rRNA --summary-file $outdir_step2_rRNA/${id}_rRNA_result.txt \\
-1 $outdir_step1_fastp/$id.r1.fq.gz -2 $outdir_step1_fastp/$id.r2.fq.gz &&
rm $outdir_step2_rRNA/${id}_accepted_hits.sam &&
mv $outdir_step2_rRNA/${id}_non_rRNA.1 $outdir_step2_rRNA/${id}_non_rRNA.1.fq.gz &&
mv $outdir_step2_rRNA/${id}_non_rRNA.2 $outdir_step2_rRNA/${id}_non_rRNA.2.fq.gz &&
Reads Alignment
hisat2 -k 10 -p 12 --no-unal \\
-x index_path -S $outdir_step3_hisat2/${id}_accepted_hits.sam \\
--novel-splicesite-outfile $outdir_step3_hisat2/${id}_junctions.bed \\
--fr --dta --avoid-pseudogene \\
--un-conc-gz $outdir_step3_hisat2/${id}_unmapped --summary-file $outdir_step3_hisat2/${id}_hisat2_result.txt \\
-1 $outdir_step2_rRNA/${id}_non_rRNA.1.fq.gz -2 $outdir_step2_rRNA/${id}_non_rRNA.2.fq.gz &&
samtools view -@ 4 -Su $outdir_step3_hisat2/${id}_accepted_hits.sam | samtools sort -@ 4 -o $outdir_step3_hisat2/${id}_accepted_hits.sorted.sam -O SAM &&
samtools view -@ 4 -bS $outdir_step3_hisat2/${id}_accepted_hits.sorted.sam > $outdir_step3_hisat2/${id}_accepted_hits.sorted.bam && samtools index $outdir_step3_hisat2/${id}_accepted_hits.sorted.bam &&
rm $outdir_step3_hisat2/${id}_accepted_hits.sam $outdir_step3_hisat2/${id}_accepted_hits.sorted.sam
Quantification
此处使用featureCounts对上一步的比对结果进行定量,速度还是非常快的。
featureCounts \
-T 12 -t gene -g gene_id -B -C -p -O --minOverlap 10 \
-a GCF_000001405.40_GRCh38.p14_genomic.gtf \
-o ${outdir_step4_featureCounts_1}/$id.featurecounts.txt ${dir}/result/$id/step3_hisat2/${id}_accepted_hits.sorted.bam
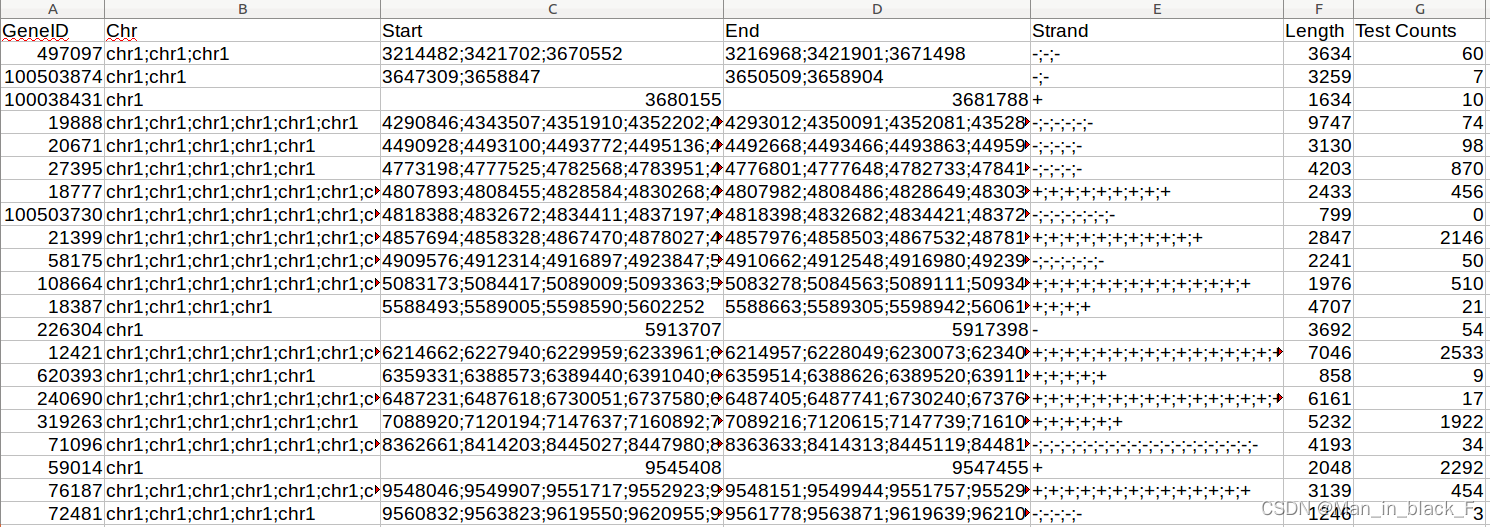
之后便可得到各基因定量的结果,但要注意的是,这里输出的计数是reads数目,需要根据自己的需要将各基因的表达量进行标准化。
这里给出各标准化的公式,大家自行选择。
其他
RPKM/FPKM
- 定义:
RPKM: Reads Per Kilobase of exon model per Million mapped reads (每千个碱基的转录每百万映射读取的reads);
FPKM: Fragments Per Kilobase of exon model per Million mapped fragments(每千个碱基的转录每百万映射读取的fragments) - 计算公式:

TPM
- 定义:
TPM: Transcripts Per Kilobase of exon model per Million mapped reads (每千个碱基的转录每百万映射读取的Transcripts) - 计算公式:

稍微解释一下:
将reads counts除以每个基因的长度(以千碱基为单位),得到每千碱基reads(RPK, reads per kilobase)。
计算样本中所有RPK值,然后将其除以1,000,000,得到“每百万”缩放因子 ( “per million”scaling factor )。
将RPK值除以“每百万”缩放因子,得到TPM。
到此为止,我们测序数据的处理就完成了,后续就是获取各个样本的表达矩阵,进行分析啦。
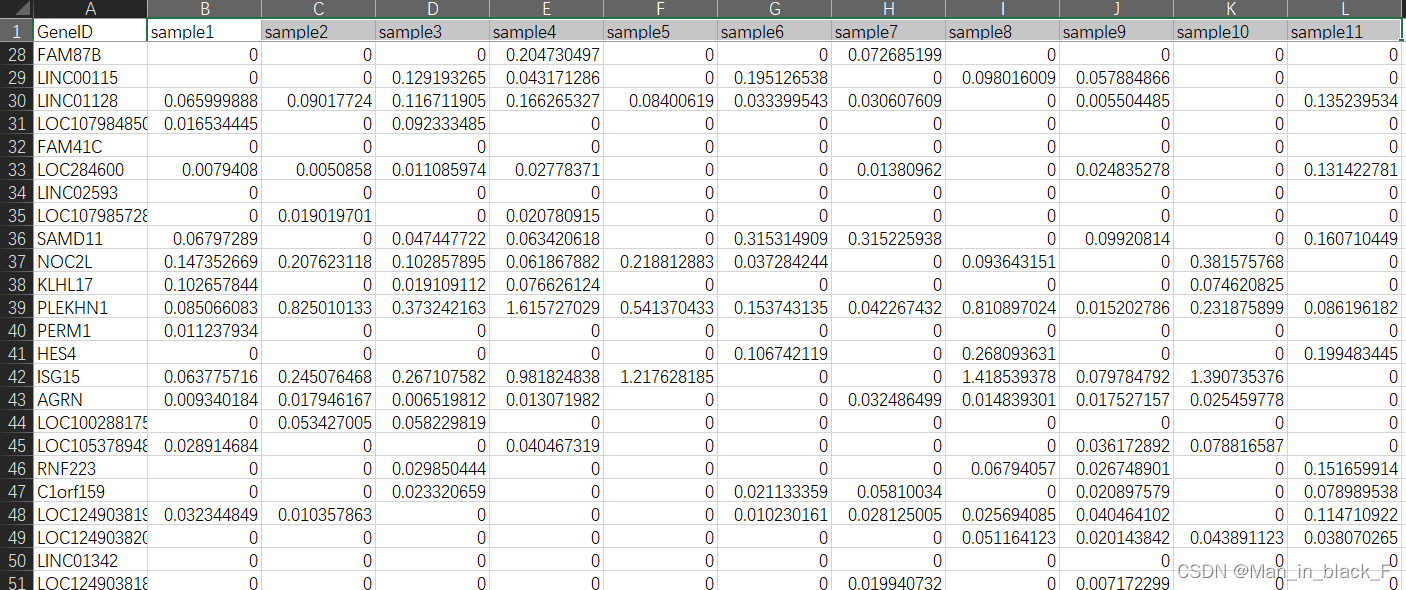
!!!(建议准备两个表达矩阵,一个是标准化后的,一个是reads数的)!!!

参考
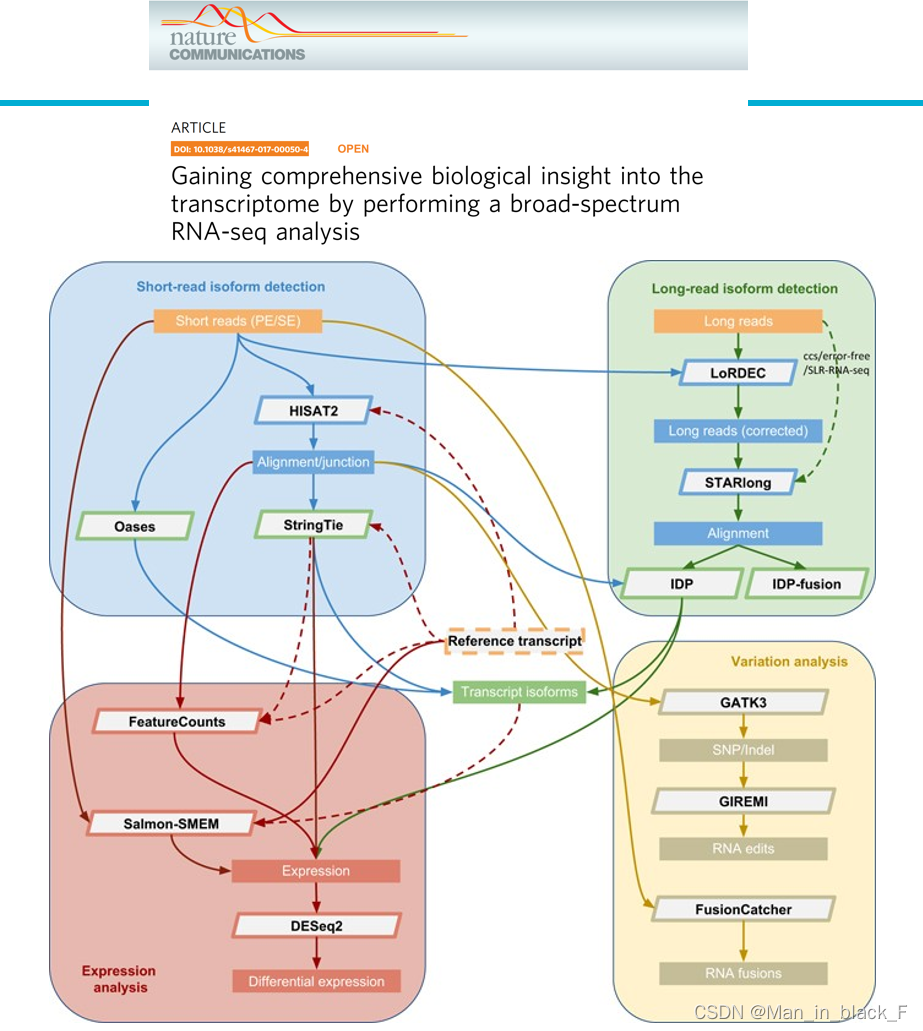
Sahraeian S M E, Mohiyuddin M, Sebra R, et al. Gaining comprehensive biological insight into the transcriptome by performing a broad-spectrum RNA-seq analysis[J]. Nature communications, 2017, 8(1): 1-15.
Conesa A, Madrigal P, Tarazona S, et al. A survey of best practices for RNA-seq data analysis[J]. Genome biology, 2016, 17(1): 1-19.
Marco-Puche G, Lois S, Benítez J, et al. RNA-Seq perspectives to improve clinical diagnosis[J]. Frontiers in genetics, 2019, 10: 1152.
RPKM、FPKM、TPM详解
Gene expression units explained: RPM, RPKM, FPKM, TPM, DESeq, TMM, SCnorm, GeTMM, and ComBat-Seq


















 3万+
3万+

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











