







什么是单细胞测序
单细胞RNA-Seq提供成千上万个单个细胞的 transcriptional profiling。这种水平的通量分析使研究人员能够在单细胞水平上了解哪些基因表达,多少数量以及异质样品中成千上万个细胞之间的差异。
目前,测序可以回答以下6类问题:
DNA的序列:ATCG怎么排列,以及各序列的丰度;
DNA的表观遗传修饰:比如甲基化、羟甲基化,以及组蛋白的各种修饰;
RNA的序列:AUCG怎么排列,以及各序列的丰度;
RNA的表观遗传修饰:比如近年很火的m6A修饰;
染色质的结构:3C、4C、5C等各种C;
其他魔性应用:比如DNA损伤位置、蛋白-蛋白相互作用等。
单细胞测序,就是想办法在单细胞层面去回答以上6类问题。
为什么要有单细胞测序
如果把这个问题换个姿势来问,那就变成,为什么非用单细胞测序不可?
世界上没有两片相同的叶子。对于多细胞生物来说,细胞与细胞之间是有差异的。当然了,这个差异可大可小。
比如说,受精卵从一个细胞开始分裂,并逐渐形成囊胚,最终发育成个体的时候,细胞与细胞之间的差异会越来越大:有的分化成神经元,有的分化成骨骼肌,各自表达着不同的遗传信息,承担着不同的生理功能。
又比如在肿瘤组织中,肿块中心的细胞,肿块周围的细胞,淋巴转移灶的细胞,以及远端转移的细胞,其基因组和转录组等遗传信息,是存在差异的。而这种差异,在临床上,可以决定该肿瘤对某种疗法是否有效。
这就是所谓的遗传信息的异质性。(异质性是遗传学概念,一种遗传性状可以由多个不同的遗传物质改变所引起。)
单细胞测序可以得到不同细胞/单个细胞的异质性信息,可以探究细胞各自表达着不同的遗传信息。
传统的研究方法,是在多细胞水平进行的。因此,最终得到的信号值,其实是多个细胞的平均,丢失了异质性的信息。
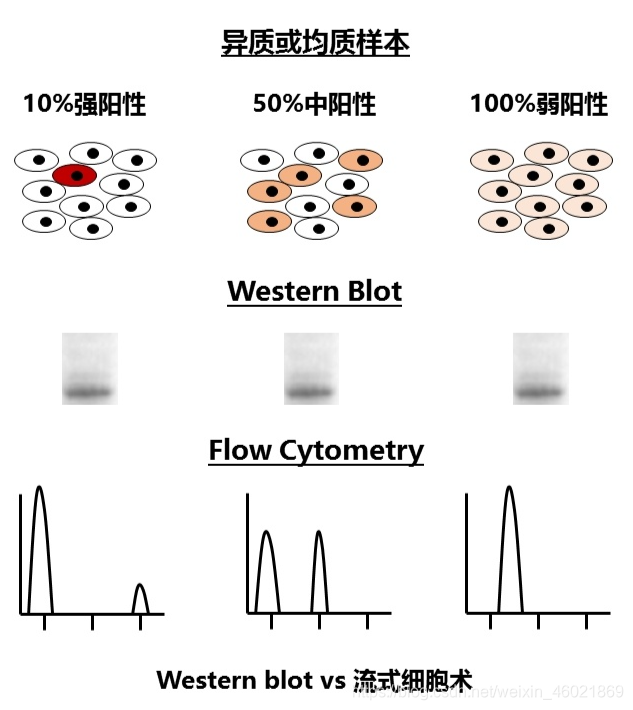
为了检测某个蛋白质的表达量,我们可以用Western blot和流式细胞术来实现。但是,用Western
blot的话,我们并没有办法区分上述的情况:目的蛋白只在10%的细胞中强表达,还是在50%的细胞里中等表达,还是在所有细胞中弱表达呢?因为最终电泳跑出来,就是一条差不多强度的带。但如果用流式细胞术这种在单细胞水平对荧光强度加以测定的技术,就能区分上述的情况了。同样道理,单细胞测序能够检出混杂样品测序所无法得到的异质性信息。而这将带领整个遗传学领域进入新的次元。
cDNA是指互补(有时称拷贝)DNA。特指在体外经过逆转录后与RNA互补的DNA链。cDNA没有内含子而只有外显子的序列
怎么做单细胞测序
方法一:根据网上介绍可以采用流式细胞仪
方法二:
基于标签(barcode)的单细胞识别。它的主要思想是,给每个细胞加上独一无二的DNA序列,这样在测序的时候,就把携带相同barcode的序列视为来自同一个细胞了。这种策略,可以通过一次建库,测得数百上千个单细胞的信息。
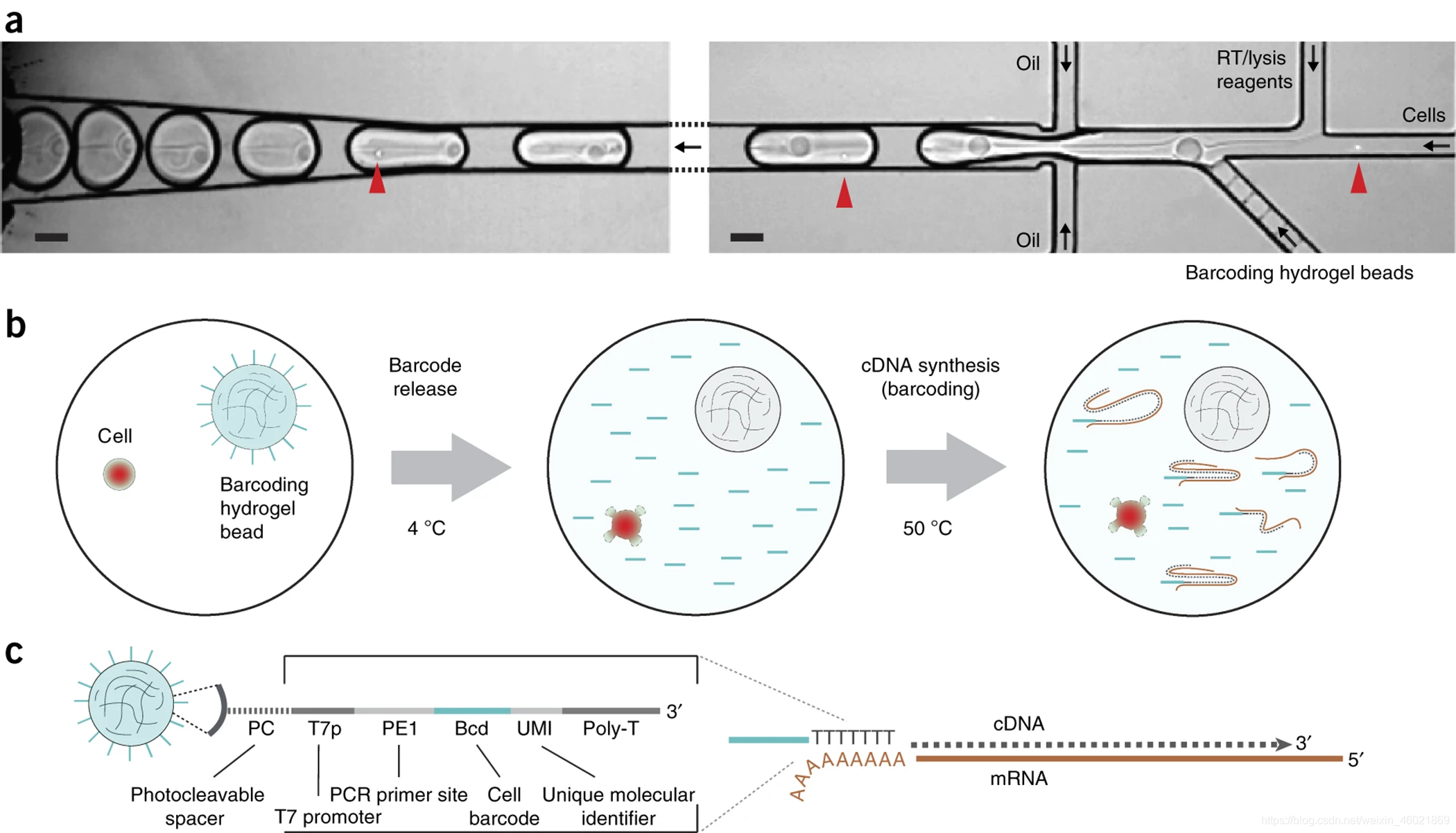
(a)将细胞,条形码(由水凝胶珠粒递送)和RT裂解试剂共封装到微流体液滴中。红色箭头表示单个单元格,黑色箭头表示流动方向。收集到所需数量的细胞后,通过光裂解从水凝胶珠中释放条形码引物,以启动mRNA的逆转录。比例尺,100μm。(b)液滴中单细胞转录组条形码的示意图。细胞和水凝胶珠粒包封后,使用>
350 nm紫外光(对DNA /
RNA无损害)从珠粒中释放条形码cDNA引物,然后进行mRNA捕获和逆转录。(c)附在水凝胶珠上的条形码cDNA引物的示意图。mRNA为棕色,而cDNA为灰色虚线。
图中部分标识解释:
barcode : 每一个单细胞的cDNA文库,就带上了独一无二的barcode了,barcode是用来区分细胞的,跟umi不同
umi:(Unique Molecular Identifiers)是短序列,用于唯一标记样品库中的每个分子。UMI被广泛用于测序应用,大多关于DNA和cDNA的PCR重复。UMI去重复还可用于RNA-seq基因表达分析和其他定量测序方法。简而言之:UMI是唯一可识别的短序列,作为umi是唯一的,所以我们可以知道在PCR扩增的过程中哪些是由同一条DNA扩增出来的。
上图的补充知识:mRNA的成熟过程
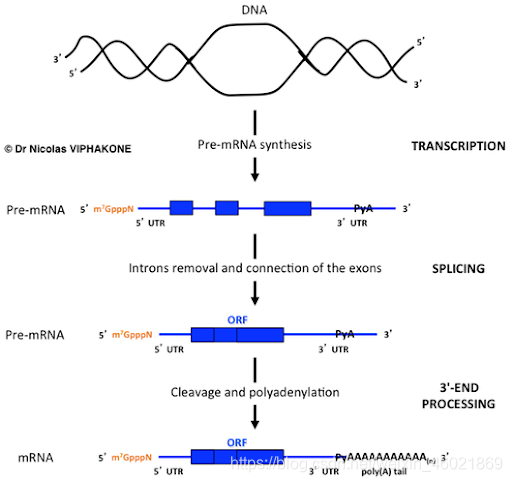 可以看到成熟的mRNA的最右边有一串"AAAAAAAA",所以包含条形码化凝胶珠的poly T那端可以配对mRNA的poly A,从而进行逆转录为cDNA。复制方向:5’ to 3’
可以看到成熟的mRNA的最右边有一串"AAAAAAAA",所以包含条形码化凝胶珠的poly T那端可以配对mRNA的poly A,从而进行逆转录为cDNA。复制方向:5’ to 3’
单细胞测序的方法
(待补充)
三种方法
单细胞测序的三种方式:https://cloud.tencent.com/developer/article/1675270
smart_seq2:全场测序
10X:一般都不是全长测序,而是3’端单细胞转录组测序
(不一定要全长测序才能测出intron)
单细胞测序分析的数据是什么数据,什么数据类型?
(待补充)
参考链接:
- 知乎:https://zhuanlan.zhihu.com/p/28844468
文 - 文献图片:https://www.rna-seqblog.com/single-cell-barcoding-and-sequencing-using-droplet-microfluidics/

















 1423
1423

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









