






来自:生物信息学-陈铭第二版的一个例题。
题目:

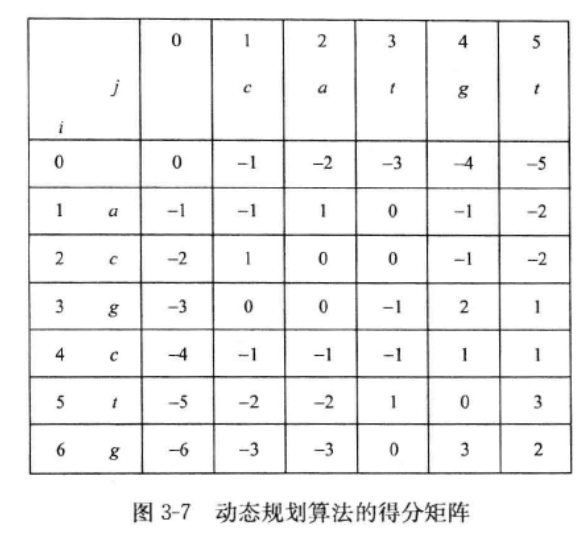

目前的代码,运行不正确,关键就是不知道怎么回溯啊,回溯怎么标记呢?


#include <iostream> #include<vector> using namespace std; vector<char> s1,t1;//在回溯的时候使用 string s,t;//输入两个字符串 int dp[30][30]; int maxs(int x,int y,int z){ if(x>=y&&x>=z)return x; else if(y>=x&&y>=z)return y; else return z; } void dfs(int len1,int len2){ //递归出口 if(len1==0||len2==0){//这里有一个问题就是另一个没放完怎么办?我要的是全部的。 //如果有一个其中没放完,那么接着放。 if(len1!=0){//如果s1没放完那么 for(int i=len1-1;i>=0;i--){ s1.push_back(s[i]); t1.push_back('-'); } } if(len2!=0){ for(int i=len2-1;i>=0;i--){ s1.push_back('-'); t1.push_back(t[i]); } } //打印s1,t1。 for(int i=s1.size()-1;i>=0;i--) cout<<s1[i]<<" "; cout<<"\n"; for(int i=t1.size()-1;i>=0;i--) cout<<t1[i]<<" "; cout<<"\n\n"; return ; } //先求出左上、上、下三者中最大的值 int m=maxs(dp[len1-1][len2-1],dp[len1-1][len2],dp[len1][len2-1]); if(dp[len1-1][len2-1]==m){//如果这样的话,那么就将二者push进来 s1.push_back(s[len1-1]); t1.push_back(t[len2-1]); dfs(len1-1,len2-1); s1.pop_back();t1.pop_back(); } if(dp[len1-1][len2]==m){ s1.push_back(s[len1-1]); t1.push_back('-'); dfs(len1-1,len2); s1.pop_back();t1.pop_back(); } if(dp[len1][len2-1]==m){ s1.push_back('-'); t1.push_back(t[len2-1]); dfs(len1,len2-1); s1.pop_back();t1.pop_back(); } } int main() { cin>>s>>t; int len1=s.size(); int len2=t.size(); int x,y,z; for(int i=0;i<=len1;i++){ for(int j=0;j<=len2;j++){ if(i==0&&j==0){ dp[i][j]=0;continue; } // if(i==0||j==0){//这么写是不对的,因为不知道是哪个插入了多少空格。 // dp[i][j]=-1;continue; // } if(i==0){//相当于在s中一直插入-。 dp[i][j]=dp[i][j-1]-1;continue; } if(j==0){ dp[i][j]=dp[i-1][j]-1;continue; } x=dp[i][j-1]-1;//在t中插入空格 y=dp[i-1][j-1]+(s[i-1]==t[j-1]?2:-1);//比较两者是否相同,相同+2,否则-1 z=dp[i-1][j]-1;//在s中插入空格 dp[i][j]=maxs(x,y,z); }//在计算y时,一开始没有加上三元表达式的括号,导致计算错误。 } //下面是怎么回溯找到解呢??? for(int i=0;i<=len1;i++){ for(int j=0;j<=len2;j++){ cout<<dp[i][j]; if(dp[i][j]<0)cout<<" "; else cout<<" "; } cout<<"\n"; } dfs(len1,len2); return 0; } /** acgctg catgt **/
目前的运行结果:
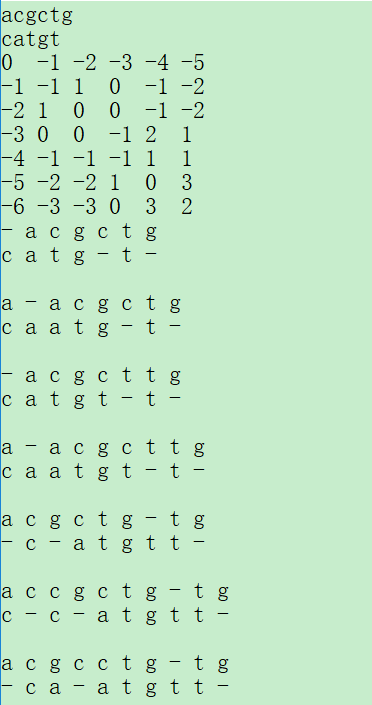
dp矩阵是正确的但是回溯结果不对,还是编程能力有限,先思考着,有解了再过来。

















 256
256

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









