



邻居列表方法(Neighbor List Method)是一种在分子动力学模拟中提高计算效率的重要技术,用于减少粒子之间相互作用的计算次数。此方法通过构建一个动态更新的邻居列表,限制计算仅在相互作用范围内的邻居之间进行,从而减少不必要的计算。
一、邻居列表方法的基本概念
在分子动力学模拟中,每个粒子与其他粒子之间的相互作用需要根据相对距离计算。然而,计算所有粒子对之间的相互作用耗费大量时间,尤其当粒子数 较大时,总的计算量为
。为了减少这种复杂度,可以设定一个截断距离
(cutoff distance),并假设超出该距离的粒子间相互作用可以忽略不计。
通过限制作用力的计算范围,仅计算邻居列表中的粒子对。这样在大多数时间步中可以避免不必要的计算,将复杂度从 降低到近似
。
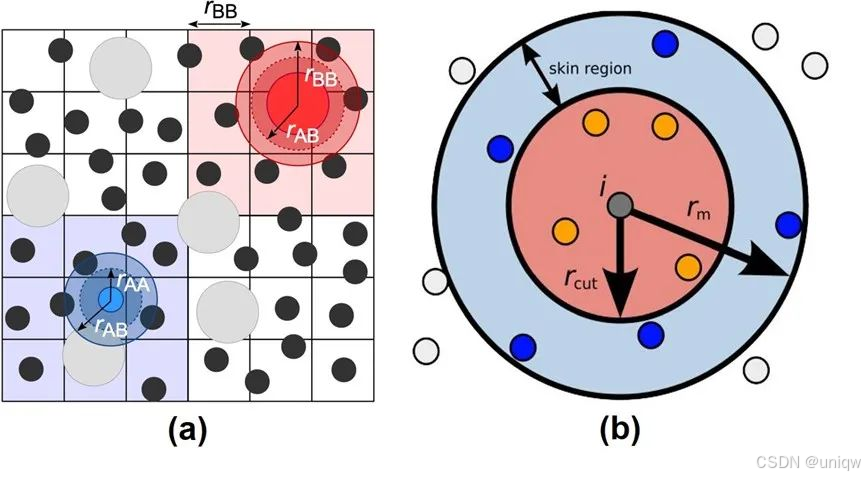
在实际操作中,通常在设定的截断距离上加上一个额外的“安全距离” 以保证邻居列表的有效性。这样可以在不频繁更新列表的情况下保证计算的精度。
二、邻居列表方法应用步骤
邻居列表方法应用步骤如下:
1.确定截断距离:设定一个最大相互作用距离 。
2.生成邻居列表:在每个时间步或间隔一定步数后,记录每个粒子在 范围内的“邻居”粒子。直接计算每个粒子与其他所有粒子的距离,选择符合距离要求的邻居。
3.限制计算范围:在相互作用计算时,仅计算在邻居列表中的粒子对。
4.定期更新:由于粒子位置随时间变化,邻居列表需要定期更新,保证计算的精确性。在每隔一定的步数更新邻居列表,避免频繁重建邻居列表带来的开销,但仍然保证计算结果的准确性。
三、邻居列表方法在分子动力学模拟中的应用
邻居列表方法在分子动力学模拟中主要应用于加速范德华力、库伦力等短程相互作用力的计算。
以上篇文章“用Python语言编写初级分子动力学模拟框架程序”为例:
代码运行速度比较局限于电脑性能。
在上述程序中对于粒子间的范德华作用力中引入邻居列表方法:
1.进行参数设定
import numpy as np
rcut = 2.5 # 截断距离
neighbor_update_interval = 10 # 更新邻居列表的时间步间隔,可根据需求更改
2.在函数定义中加入创建邻居列表的代码模块
# 创建邻居列表
def create_neighbor_list(positions):
neighbor_list = []
for i in range(N):
disp = apply_pbc(positions - positions[i])
r2 = np.sum(disp ** 2, axis=1)
neighbors = np.where((r2 < (rcut + skin) ** 2) & (r2 > 0))[0]
neighbor_list.append(neighbors)
return neighbor_list
3.修改主程序以调用邻居列表
# 主程序
positions, velocities = initialize_particles()
neighbor_list = create_neighbor_list(positions)
forces = calculate_forces(positions, neighbor_list)
write_gro_file("initial_structure.gro", positions)
for step in range(time_steps):
if step % neighbor_update_interval == 0:
neighbor_list = create_neighbor_list(positions)
positions, velocities, forces = velocity_verlet(positions, velocities, forces, neighbor_list)
velocities = rescale_velocities(velocities, k_B_T)
write_gro_file("final_structure.gro", positions)引入邻居列表方法方法后,代码运行速度显著上升,相比于之前提高近6倍。
四、邻居列表方法的优点与局限性
优点:邻居列表法显著减少了计算时间,提高了大规模分子系统模拟的可行性和效率。
局限性:邻居列表构建和更新本身也消耗一定计算资源;此外,设定的截断距离可能忽略某些远距离作用力,影响模拟精度。
通过邻居列表方法,分子动力学模拟可以处理更大规模和更长时间的系统,显著提高了资源利用效率。这一方法广泛应用于物理、化学和生物分子模拟中,尤其在模拟包含数十万到数百万粒子的复杂体系时,邻居列表方法至关重要。


















 1303
1303

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









