







人群分层:一项研究中存在多个亚群(例如具有不同种族背景的个体),因为等位基因频率在亚群之间可能不同,所以群体分层可能导致假阳性关联和/或掩盖真实关联。例如筷子基因,由于人群分层导致一个SNP占了使用筷子能力的变异的一半。
一般来说,如果样本包括多个种族,建议分别对每个种族进行关联测试,并使用适当方法将结果进行联合。
GWAS系统偏差的一个重要来源是人口分层。
plink中使用MDS方法矫正人群分层,该方法计算样本中任何一对个体之间共享的全基因组平均等位基因比例,以生成每个个体遗传变异的定量指数,可以绘制个体成分分数以探索是否存在遗传上比预期更相似的个体群体。例如,在一项包括来自亚洲和欧洲的受试者的基因研究中,MDS分析将揭示亚洲人在基因上比欧洲人更相似。
多维尺度分析(Mutidimensional scaling-MDS)的目标是将对象间的差异(或相似性)通过图形展示出来,图中两点的距离表示对象差异。距离越远表示两个对象差异越大,距离越近表示两个对象差异越小。空间图的最表不一定有实际意义。
基于MDS分析的异常值个体应从进一步分析中删除,排除这些个体后,必须进行新的MDS分析。其主要成分需要作用关联分析中的协变量,以校正人群中任何剩余的人群分层,需要包含多少成分取决于人口结构和样本量,但进行遗传学界普遍接受最多包含10个成分。
在本教程中,我们将检查人口分层。
在上一教程结束时生成的bfile文件(HapMap_3_r3_12)将使用来自1000基因组计划的数据检查人群分层。非欧洲种族背景的个人将被移除。
此外本教程将生成一个协变量文件,以帮助调整欧洲受试者的剩余人口分层。
Download 1000 Genomes data
这个来自1000个基因组的文件包含了来自不同种族背景的629个个体的基因数据。
#注意,这个文件相当大(> 60G)。
Wget ftp://ftp-trace.ncbi.nih.gov/1000genomes/ftp/release/20100804/ALL.2of4intersection.20100804.genotypes.vcf.gz
Convert vcf to Plink format.
plink --vcf ALL.2of4intersection.20100804.genotypes.vcf.gz --make-bed --out ALL.2of4intersection.20100804.genotypes

 需要注意的是,ALL.2of4intersection.20100804.genotypes.bim包含的SNP没rs标识符,这些SNPs用“.”表示。这个也可以在ALL.2of4intersection.20100804.genotypes.vcf.gz文件中发现。
需要注意的是,ALL.2of4intersection.20100804.genotypes.bim包含的SNP没rs标识符,这些SNPs用“.”表示。这个也可以在ALL.2of4intersection.20100804.genotypes.vcf.gz文件中发现。
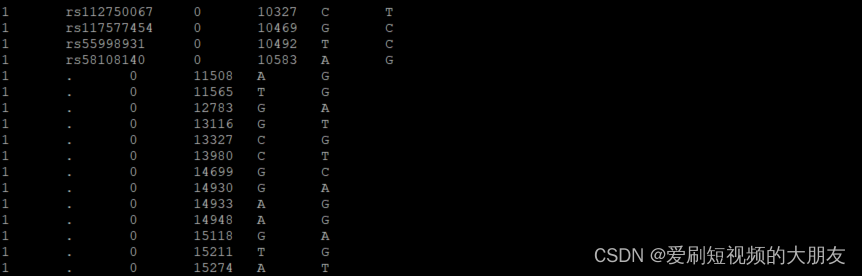
在1000基因组数据中缺少rs-标识符在本教程中不是问题。
#然而,作为良好的实践,我们将为缺失rs-标识符的snp分配唯一标识符(即带有“.”的snp)。
plink --bfile ALL.2of4intersection.20100804.genotypes --set-missing-var-ids @:#[b37]\$1,\$2 --make-bed --out ALL.2of4intersection.20100804.genotypes_no_missing_IDs

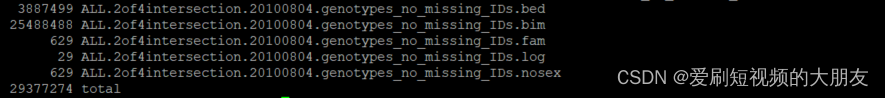
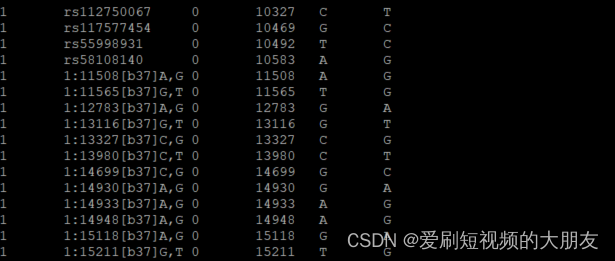 可以看到,上个代码为每个没有名字的SNP都分配了唯一的标识符用以区分。
可以看到,上个代码为每个没有名字的SNP都分配了唯一的标识符用以区分。
对该数据进行质控
## QC on 1000 Genomes data.
# Remove variants based on missing genotype data.
plink --bfile ALL.2of4intersection.20100804.genotypes_no_missing_IDs --geno 0.2 --allow-no-sex --make-bed --out 1kG_MDS
# Remove individuals based on missing genotype data.
plink --bfile 1kG_MDS --mind 0.2 --allow-no-sex --make-bed --out 1kG_MDS2
# Remove variants based on missing genotype data.
plink --bfile 1kG_MDS2 --geno 0.02 --allow-no-sex --make-bed --out 1kG_MDS3
# Remove individuals based on missing genotype data.
plink --bfile 1kG_MDS3 --mind 0.02 --allow-no-sex --make-bed --out 1kG_MDS4
# Remove variants based on MAF.
plink --bfile 1kG_MDS4 --maf 0.05 --allow-no-sex --make-bed --out 1kG_MDS5
#从1000个基因组数据集中提取HapMap数据集中的变体。
awk '{print$2}' HapMap_3_r3_12.bim > HapMap_SNPs.txt
plink --bfile 1kG_MDS5 --extract HapMap_SNPs.txt --make-bed --out 1kG_MDS6
 HapMap_SNPs.txt文件一共1073226行。
HapMap_SNPs.txt文件一共1073226行。
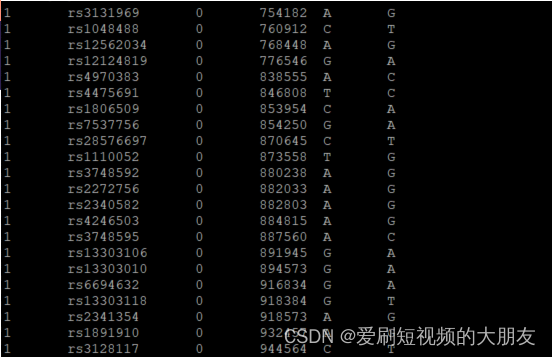 1kG_MDS6.bim文件一共1000993行,查看文件表明
1kG_MDS6.bim文件一共1000993行,查看文件表明--extract命令将HapMap_SNPs.txt文件中提取1kG_MDS5.bim文件中存在的SNP。
#从HapMap数据集中提取1000基因组数据集中的变体。
awk '{print$2}' 1kG_MDS6.bim > 1kG_MDS6_SNPs.txt
plink --bfile HapMap_3_r3_12 --extract 1kG_MDS6_SNPs.txt --recode --make-bed --out HapMap_MDS
HapMap_MDS.bim文件中包含1000993行数据。
#数据集现在包含完全相同的变体。
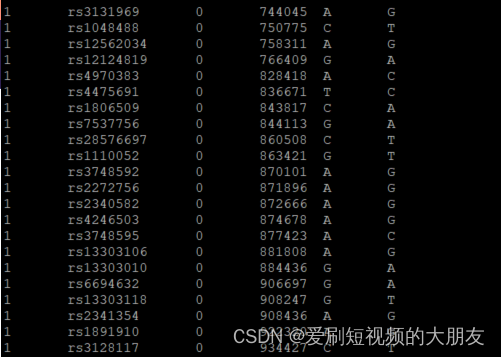
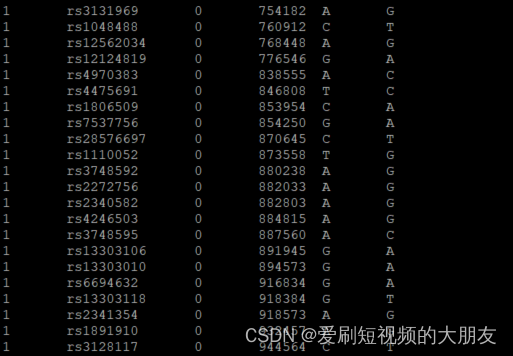 可以看到,HapMap_MDS.bim和1kG_MDS6.bim文件具有相同的的SNP,但是由于使用的参考基因组不同,第四列的物理位置不同,需要将这两个文件进行统一。
可以看到,HapMap_MDS.bim和1kG_MDS6.bim文件具有相同的的SNP,但是由于使用的参考基因组不同,第四列的物理位置不同,需要将这两个文件进行统一。
awk '{print$2,$4}' HapMap_MDS.map > buildhapmap.txt
#buildmap .txt每行包含一个SNP-id和物理位置。
plink --bfile 1kG_MDS6 --update-map buildhapmap.txt --make-bed --out 1kG_MDS7
#1kG_MDS7和HapMap_MDS现在具有相同的构建。
合并HapMap和1000个基因组数据集
#1) set reference genome
awk '{print$2,$5}' 1kG_MDS7.bim > 1kg_ref-list.txt
plink --bfile HapMap_MDS --reference-allele 1kg_ref-list.txt --make-bed --out HapMap-adj
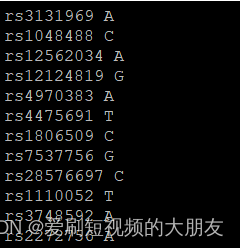 1kg_ref-list.txt包含1kG_MDS7.bim第二列的SNP名称和第五列的参考等位基因。
1kg_ref-list.txt包含1kG_MDS7.bim第二列的SNP名称和第五列的参考等位基因。
转换前
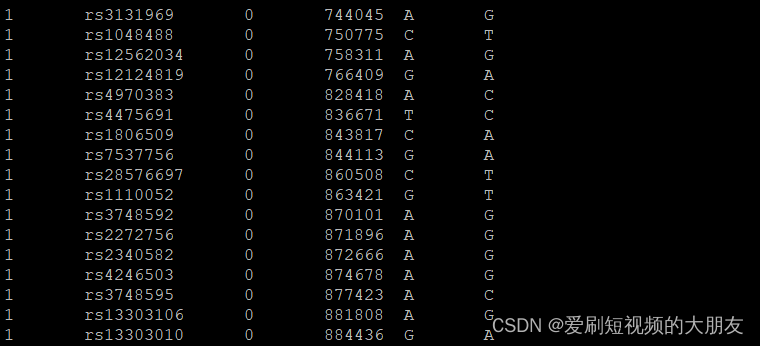 转换后
转换后
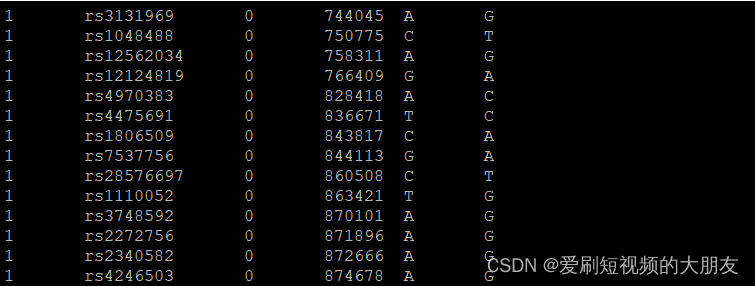 可以看到两个文件的第十行的rs1110052位点的两个等位基因调换了位置,这是因为–reference-allele命令以1kg_ref-list.txt中的信息为参考等位基因,而不是以计算出的优势等位基因做为参考等位基因。
可以看到两个文件的第十行的rs1110052位点的两个等位基因调换了位置,这是因为–reference-allele命令以1kg_ref-list.txt中的信息为参考等位基因,而不是以计算出的优势等位基因做为参考等位基因。
#对于所有snp, 1kG_MDS7和hamap -adj具有相同的参考基因组。
#该命令将为不可能的A1等位基因分配产生一些警告。
awk '{print$2,$5,$6}' 1kG_MDS7.bim > 1kGMDS7_tmp
less 1kG_MDS7.bim
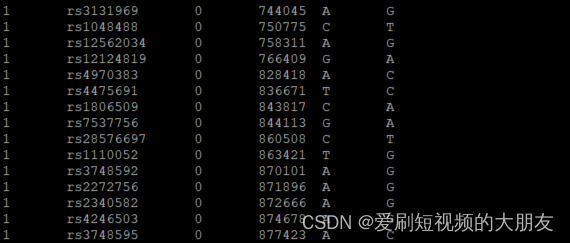
less 1kGMDS7_tmp
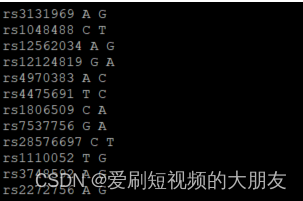
awk '{print$2,$5,$6}' HapMap-adj.bim > HapMap-adj_tmp
less HapMap-adj.bim
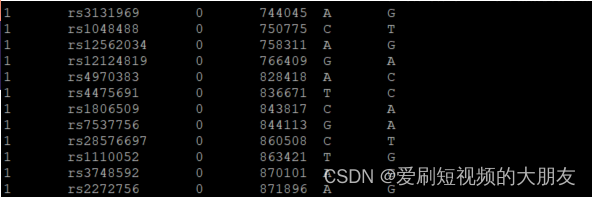
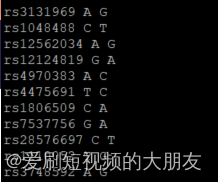
sort 1kGMDS7_tmp HapMap-adj_tmp |uniq -u > all_differences.txt
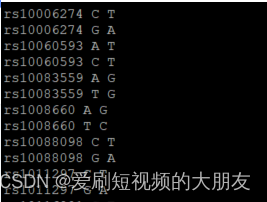 sort命令:对文本文件的第一列以ASCII码进行排序。
sort命令:对文本文件的第一列以ASCII码进行排序。
uniq命令:删除文件中的重复行。-u选项:仅显示不重复的行。
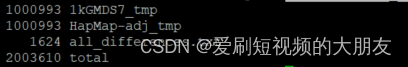
wc -l 1kGMDS7_tmp HapMap-adj_tmp all_differences.txt
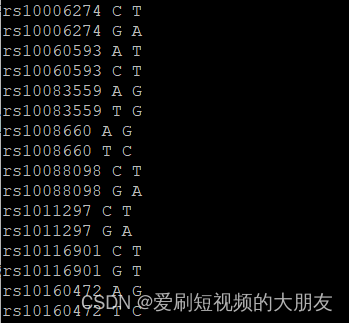
可以看到,该文件中的SNP是两个相同的SNP一组,只是这两个相同的SNP具有的ref和ale正好互补,这是因为它们被分别注释到了两条链上。
#将这些有问题的SNP去重后提取出来。
awk '{print$1}' all_differences.txt | sort -u > flip_list.txt
#Generates a file of 812 SNPs. These are the non-corresponding SNPs between the two files.
将提取出来的SNP根据1kg_ref-list.txt进行纠正。
plink --bfile HapMap-adj --flip flip_list.txt --reference-allele 1kg_ref-list.txt --make-bed --out corrected_hapmap
再检查一下是否还有不同的SNP。
awk '{print$2,$5,$6}' corrected_hapmap.bim > corrected_hapmap_tmp
sort 1kGMDS7_tmp corrected_hapmap_tmp |uniq -u > uncorresponding_SNPs.txt
uncorresponding_SNPs.txt文件表明人有84处不同(分别由42个SNP导致)。
直接将这些SNP移除。
awk '{print$1}' uncorresponding_SNPs.txt | sort -u > SNPs_for_exlusion.txt
plink --bfile corrected_hapmap --exclude SNPs_for_exlusion.txt --make-bed --out HapMap_MDS2
plink --bfile 1kG_MDS7 --exclude SNPs_for_exlusion.txt --make-bed --out 1kG_MDS8
现在可以将这两个文件进行合并了。
plink --bfile HapMap_MDS2 --bmerge 1kG_MDS8.bed 1kG_MDS8.bim 1kG_MDS8.fam --allow-no-sex --make-bed --out MDS_merge2
Perform MDS on HapMap-CEU data anchored by 1000 Genomes data.
plink --bfile MDS_merge2 --extract indepSNP.prune.in --genome --out MDS_merge2
plink --bfile MDS_merge2 --read-genome MDS_merge2.genome --cluster --mds-plot 10 --out MDS_merge2
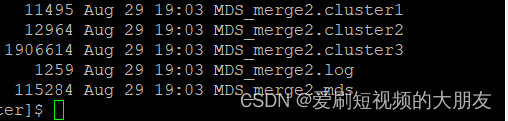
最终生成了这些文件。
接着就是将这个MDS分析结果进行可视化。
在这之前,需要下载以下1000基因组文件中每个个体对应的种群代码,然后将种群代码更改一下。
wget ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/release/20100804/20100804.ALL.panel
awk '{print$1,$1,$2}' 20100804.ALL.panel > race_1kG.txt
sed 's/JPT/ASN/g' race_1kG.txt>race_1kG2.txt
sed 's/ASW/AFR/g' race_1kG2.txt>race_1kG3.txt
sed 's/CEU/EUR/g' race_1kG3.txt>race_1kG4.txt
sed 's/CHB/ASN/g' race_1kG4.txt>race_1kG5.txt
sed 's/CHD/ASN/g' race_1kG5.txt>race_1kG6.txt
sed 's/YRI/AFR/g' race_1kG6.txt>race_1kG7.txt
sed 's/LWK/AFR/g' race_1kG7.txt>race_1kG8.txt
sed 's/TSI/EUR/g' race_1kG8.txt>race_1kG9.txt
sed 's/MXL/AMR/g' race_1kG9.txt>race_1kG10.txt
sed 's/GBR/EUR/g' race_1kG10.txt>race_1kG11.txt
sed 's/FIN/EUR/g' race_1kG11.txt>race_1kG12.txt
sed 's/CHS/ASN/g' race_1kG12.txt>race_1kG13.txt
sed 's/PUR/AMR/g' race_1kG13.txt>race_1kG14.txt
将自己的文件中的个个单独赋予一个代码。
awk '{print$1,$2,"OWN"}' HapMap_MDS.fam>racefile_own.txt
将以上两个文件联合起来。
cat race_1kG14.txt racefile_own.txt | sed -e '1i\FID IID race' > racefile.txt
然后执行R代码进行可视化。
data<- read.table(file="MDS_merge2.mds",header=TRUE)
race<- read.table(file="racefile.txt",header=TRUE)
datafile<- merge(data,race,by=c("IID","FID"))
head(datafile)
pdf("MDS.pdf",width=7,height=7)
for (i in 1:nrow(datafile))
{
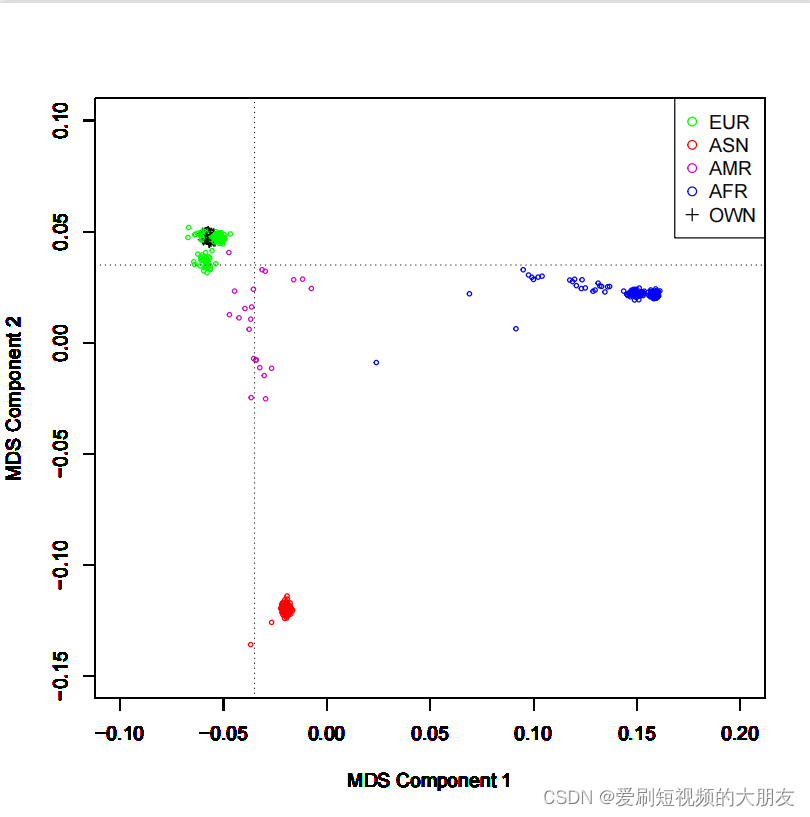
if (datafile[i,14]=="EUR") {plot(datafile[i,4],datafile[i,5],type="p",xlim=c(-0.1,0.2),ylim=c(-0.15,0.1),xlab="MDS Component 1",ylab="MDS Component 2",pch=1,cex=0.5,col="green")}
par(new=T)
if (datafile[i,14]=="ASN") {plot(datafile[i,4],datafile[i,5],type="p",xlim=c(-0.1,0.2),ylim=c(-0.15,0.1),xlab="MDS Component 1",ylab="MDS Component 2",pch=1,cex=0.5,col="red")}
par(new=T)
if (datafile[i,14]=="AMR") {plot(datafile[i,4],datafile[i,5],type="p",xlim=c(-0.1,0.2),ylim=c(-0.15,0.1),xlab="MDS Component 1",ylab="MDS Component 2",pch=1,cex=0.5,col=470)}
par(new=T)
if (datafile[i,14]=="AFR") {plot(datafile[i,4],datafile[i,5],type="p",xlim=c(-0.1,0.2),ylim=c(-0.15,0.1),xlab="MDS Component 1",ylab="MDS Component 2",pch=1,cex=0.5,col="blue")}
par(new=T)
if (datafile[i,14]=="OWN") {plot(datafile[i,4],datafile[i,5],type="p",xlim=c(-0.1,0.2),ylim=c(-0.15,0.1),xlab="MDS Component 1",ylab="MDS Component 2",pch=3,cex=0.7,col="black")}
par(new=T)
}
abline(v=-0.035,lty=3)
abline(h=0.035,lty=3)
legend("topright", pch=c(1,1,1,1,3),c("EUR","ASN","AMR","AFR","OWN"),col=c("green","red",470,"blue","black"),bty="o",cex=1)
dev.off()
命令详解
Update variant information
全外显子和全基因组测序结果频繁地包含没有标准ID的变体。如果你不想舍弃这些数据,你通常会想给它们分配基于染色体和位置的id。
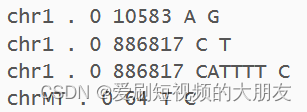
--set-missing-var-ids提供一个这样一个方式。这些标志的参数是一个特殊的模板字符串,这个命令后面可以选择两个输入模板,一个是--set-missing-var-ids @:#[b37],另一个是--set-missing-var-ids @:#[b37]\$1,\$2。例如,对于以下这个信息
 使用
使用--set-missing-var-ids @:#[b37]命令将在图中数据的第二列的第一行至第四行生成chr1:10583[b37],chr1:886817[b37],chr1:886817[b37],chrMT:64[b37].注意:有些突变体可能在同一个起始的物理位置上(例如图中的第二行和第三行),这就导致该命令在生成SNP标识符的时候会出现两个同样的SNP标识符(不符合名称的唯一性原则)。使用--set-missing-var-ids @:#[b37]\$1,\$2可以避免上述问题,在生成名称时,会加入突变体的详细信息,生成 ‘chr1:10583[b37]A,G’,‘chr1:886817[b37]C,T’,‘chr1:886817[b37]C,CATTTT’,‘chrMT:64[b37]C,T’。
但是对于包含indel的突变体,使用上述命令可能还是会生成两个相同的名称,对此需要使用PLINK2软件的--set-{all,missing}-var-ids或者bcftools,支持基于REF/ alt的命名模板。
与indel相关的等位基因名成可能非常长,而由这种长等位基因产生的合成变异ID名称,使用起来非常不方便。因此,当一个等位基因名称超过23个字符时,它会在由--set-missing-var-ids生成的变体ID中被自动截断。你可以使用--new-id-max-allele-len来改变限制。
如果你的管道没有使用’.’表示未命名的变量,你可以使用--missing-var-code指定一个不同的字符串来匹配。例如,--missing-var-code NA可以对bim文件进行填充。
因为数据中没有性别信息,所以需要使用参数--allow-no-sex以使程序允许处理没有性别信息的数据。

















 4877
4877

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









