



16S rRNA 高通量测序方法
-
样本采集与 DNA 提取
本研究的样本来源于 (描述样本来源,如肠道、土壤、唾液等),采集后迅速置于 -80°C 超低温冰箱保存,避免微生物组成变化。DNA 提取采用 MoBio PowerSoil DNA Isolation Kit(MoBio Laboratories, Carlsbad, CA, USA) 或 Qiagen DNeasy PowerSoil Kit(Qiagen, Germany),按照试剂盒说明书操作。DNA 提取的质量通过 NanoDrop 2000(Thermo Fisher Scientific, USA) 测定 A260/A280 比值(1.8~2.0 为合格),同时使用 Qubit 3.0(Invitrogen, USA) 进行荧光定量测定,并在 1% 琼脂糖凝胶上进行电泳检测 DNA 完整性。 -
16S rRNA 基因扩增
使用细菌通用引物扩增 16S rRNA 基因的 V3-V4 高变区,引物序列如下:
正向引物(Forward primer, 341F):5’-CCTAYGGGRBGCASCAG-3’
反向引物(Reverse primer, 806R):5’-GGACTACNNGGGTATCTAAT-3’
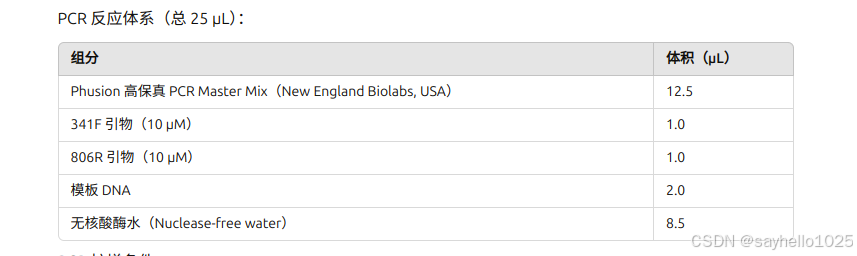
PCR 扩增条件:
95°C 预变性 3 min
95°C 变性 30 s
55°C 退火 30 s
72°C 延伸 45 s
30 个循环(steps 2-4)
72°C 终延伸 5 min
PCR 产物在 **2% 琼脂糖凝胶电泳(120V,40 min)**上检测,并使用 QIAquick PCR Purification Kit(Qiagen, Germany) 纯化。
- 测序文库构建与高通量测序
纯化后的 PCR 产物使用 NEBNext® Ultra™ DNA Library Prep Kit for Illumina®(New England Biolabs, USA) 进行测序文库构建。建库过程中,先采用 A-T 连接适配子(adapter ligation),然后利用 AMPure XP 磁珠(Beckman Coulter, USA) 进行片段筛选。文库质量评估使用 Agilent 2100 生物分析仪(Agilent Technologies, USA) 和 Qubit dsDNA HS Assay Kit(Invitrogen, USA) 进行浓度测定。
文库构建完成后,在 Illumina NovaSeq 6000 或 MiSeq PE250/PE300 平台(Illumina, USA) 进行 250 bp 或 300 bp 双端测序(paired-end sequencing),由商业测序公司(如 Novogene、BGI、Personalbio)完成。
- 原始数据处理
测序生成的原始数据(raw reads)以 FASTQ 格式存储,利用 QIIME2(v2022.8) 进行质量控制和后续分析。
去除低质量数据:
使用 Cutadapt(v1.9.1) 去除接头(adapter)污染序列;
采用 DADA2(Divisive Amplicon Denoising Algorithm, v1.18) 进行噪声过滤,去除低质量碱基、嵌合体(chimera),并去除单一出现的序列(singleton)。
拼接(Merge Paired-end Reads):
利用 VSEARCH(v2.15.2) 拼接双端序列,参数设定为 --fastq_mergepairs --fastq_minovlen 20 --fastq_maxdiffs 5。
非冗余特征表(ASV, Amplicon Sequence Variant)构建:
采用 DADA2 生成 ASV(替代传统的 OTU 操作分类单元),提高物种分类分辨率。
物种注释(Taxonomic Assignment):
物种注释采用 SILVA 138 或 Greengenes 数据库,使用 Naïve Bayes 分类器 进行比对。
5. 统计分析与可视化
Alpha 多样性(Alpha diversity):
计算 Shannon 指数、Chao1 指数、Simpson 指数(用于测定样本内微生物群落的丰富度和均匀度)。
使用 Wilcoxon 秩和检验(Wilcoxon rank-sum test) 评估组间差异。
Beta 多样性(Beta diversity):
计算 Bray-Curtis 距离、Unweighted UniFrac 和 Weighted UniFrac 距离矩阵,评估样本间微生物组成差异。
使用 主坐标分析(PCoA) 和 非度量多维缩放分析(NMDS) 进行可视化。
组间差异分析:
使用 LEfSe(Linear discriminant analysis Effect Size) 识别显著差异菌群(LDA Score > 2)。
采用 DESeq2(R 语言包) 进行差异丰度分析,筛选出显著差异菌(p < 0.05, FDR 校正)。
功能预测:
利用 PICRUSt2(Phylogenetic Investigation of Communities by Reconstruction of Unobserved States, v2.4.2) 预测微生物群落的潜在功能,分析 KEGG 和 MetaCyc 代谢通路。
6. 结果展示
所有统计分析和可视化图表均在 R 语言(v4.1.2) 和 Python 3.8 环境下完成,使用 ggplot2、phyloseq、vegan、pheatmap 等 R 包进行可视化,包括:
Alpha 和 Beta 多样性分析(箱线图、PCoA/NMDS 散点图)。
物种相对丰度柱状图(Phylum、Genus 级别)。
差异分析火山图(Volcano plot)。
LEfSe 线性判别分析柱状图。
结果解释
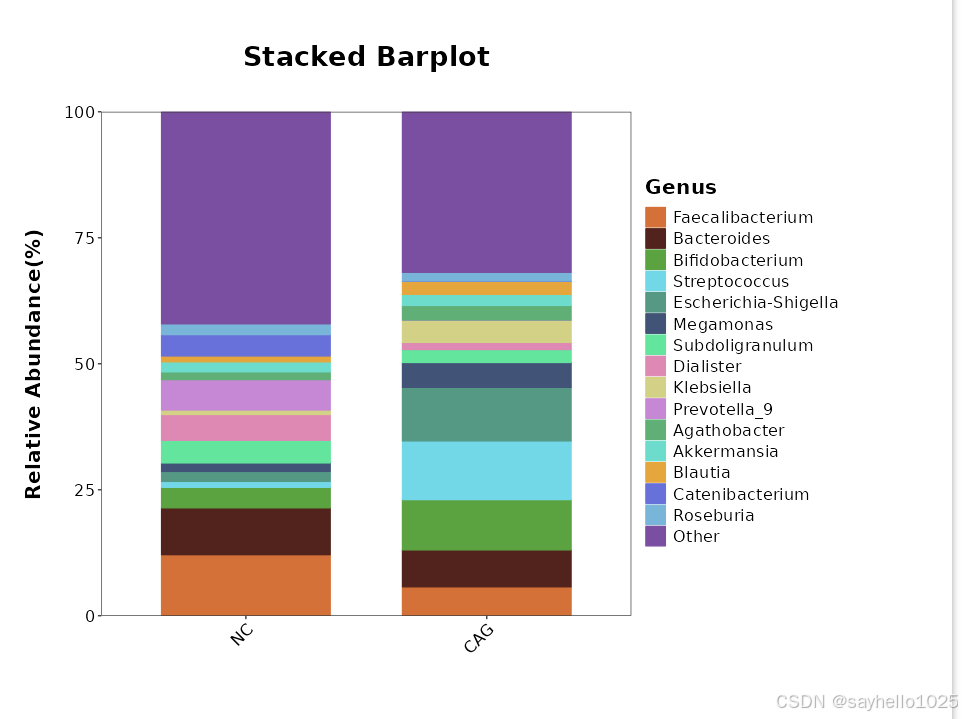
(1) NC 组富集的菌群
Faecalibacterium(粪杆菌属)
该属属于厚壁菌门(Firmicutes),是短链脂肪酸(SCFA) 生产者,特别是 丁酸(Butyrate)。
益生菌作用:具有抗炎功能,可维持肠道屏障稳定性,减少胃肠道炎症。
CAG 组减少 可能表明 抗炎菌群下降,胃肠道炎症增加。
Bacteroides(拟杆菌属)
该属属于拟杆菌门(Bacteroidota),是健康肠道中的主要共生菌群之一。
具有 参与脂肪酸和胆汁酸代谢的功能,可以分解复杂的碳水化合物并促进脂肪酸合成。
CAG 组减少 可能意味着脂肪酸生物合成减少,影响宿主能量代谢。
Bifidobacterium(双歧杆菌属)的基本特征
属于 放线菌门(Actinobacteria),是一类革兰氏阳性、厌氧或兼性厌氧的益生菌。
主要定植在肠道(尤其是婴儿期)和胃肠道上部,并在维持肠道稳态、调节免疫 方面发挥重要作用。
其主要代谢特点:
糖代谢强:通过发酵糖类(如乳糖、低聚糖)产生乳酸和乙酸,但不产生丁酸(与 Faecalibacterium 不同)。
竞争性抑制病原菌,提高胃肠道屏障功能。
与宿主免疫系统交互,可能在炎症环境中调整自身代谢方式。
(1) 胃酸减少,使双歧杆菌更易定植
正常情况下,胃酸(pH≈2)会抑制大多数外源菌在胃部的定植,但在 CAG 组,由于胃酸分泌减少,胃 pH 升高(≈4~5),这可能为 Bifidobacterium 提供更有利的生存环境。
其他乳酸菌(如 Streptococcus)也在 CAG 组增加,表明胃肠道的菌群向 糖代谢优势菌群 偏移。
(2) 肠道炎症状态改变菌群生态位
慢性萎缩性胃炎(CAG)通常伴随胃肠道低度炎症,宿主的 免疫系统改变 可能影响肠道菌群结构:
Bifidobacterium 具有一定的抗炎作用,可能通过竞争性排斥炎症相关菌(如 Proteobacteria)在 CAG 组中扩增。
在炎症环境中,Bifidobacterium 可能适应性扩增,以维持一定程度的菌群平衡。
(3) CAG 组碳水化合物代谢增强,适合 Bifidobacterium 生长
Bifidobacterium 主要代谢寡糖和糖类,而非脂肪酸,其在 CAG 组中的增加可能反映 碳水化合物代谢菌群的富集:
Faecalibacterium(SCFA 生成菌)减少,导致脂肪酸生物合成下降,而糖酵解代谢增强。
Streptococcus 也富集,进一步支持糖代谢增强的假设。
(4) CAG 可能影响胆汁酸代谢,促进双歧杆菌扩增
胆汁酸可调节肠道菌群结构:
Bifidobacterium 通常耐受低胆汁酸浓度,在胆汁酸代谢异常的情况下可能相对扩增。
CAG 可能导致胆汁酸代谢紊乱,进而影响特定菌群的竞争生态位,使 Bifidobacterium 得以扩增。
Bifidobacterium 在 CAG 组增加的可能影响
(1) 可能对宿主产生部分保护作用
双歧杆菌的抗炎作用 可能在 CAG 组中起到一定的保护作用:
竞争性抑制炎症相关菌(如 Escherichia-Shigella, Klebsiella),防止肠道菌群进一步恶化。
可能增强宿主免疫调节能力,缓解 CAG 相关的胃肠道炎症。
(2) 可能与短链脂肪酸(SCFA)失衡相关
Bifidobacterium 主要生成乳酸和乙酸,而非丁酸:
在 NC 组,Faecalibacterium(丁酸生成菌)更丰富,有助于脂肪酸生物合成。
在 CAG 组,Bifidobacterium 富集,可能导致 SCFA 组成改变,影响脂肪酸代谢。
(3) 可能与糖代谢增强相关
Bifidobacterium 在 CAG 组扩增可能表明菌群代谢向糖代谢倾斜,而非脂肪酸代谢:
链球菌(Streptococcus)和变形菌门(Proteobacteria)在 CAG 组富集,说明 CAG 组更倾向于糖酵解代谢。
Bifidobacterium 可能在这一代谢背景下扩增,以适应糖代谢环境。
CAG 组富集的菌群
Streptococcus(链球菌属)
CAG 组显著升高,可能是由于 胃酸分泌减少(胃 pH 升高),使口腔细菌更容易定植于胃部和小肠。
代谢特点:链球菌主要依靠糖酵解产生乳酸,而不参与脂肪酸生物合成。
影响:
促进炎症反应,可能加重胃黏膜损伤。
可能减少短链脂肪酸(SCFA)的产生,进一步影响宿主代谢稳态。
Escherichia-Shigella(大肠杆菌-志贺氏菌属)
该属属于 变形菌门(Proteobacteria),通常与炎症性疾病(如 IBD、胃炎)相关。
CAG 组中明显升高,可能意味着 肠道屏障功能受损,促炎菌群扩增。
Klebsiella(克雷伯氏菌属)
也是 变形菌门(Proteobacteria) 的成员,与 抗生素耐药性、宿主免疫失衡 相关。
CAG 组升高 可能表明 炎症环境有利于条件致病菌扩增。
Megamonas(巨单胞菌属)
属于厚壁菌门(Firmicutes),通常与糖代谢增强相关。
CAG 组增加 可能意味着菌群代谢偏向糖酵解,而非脂肪酸合成。
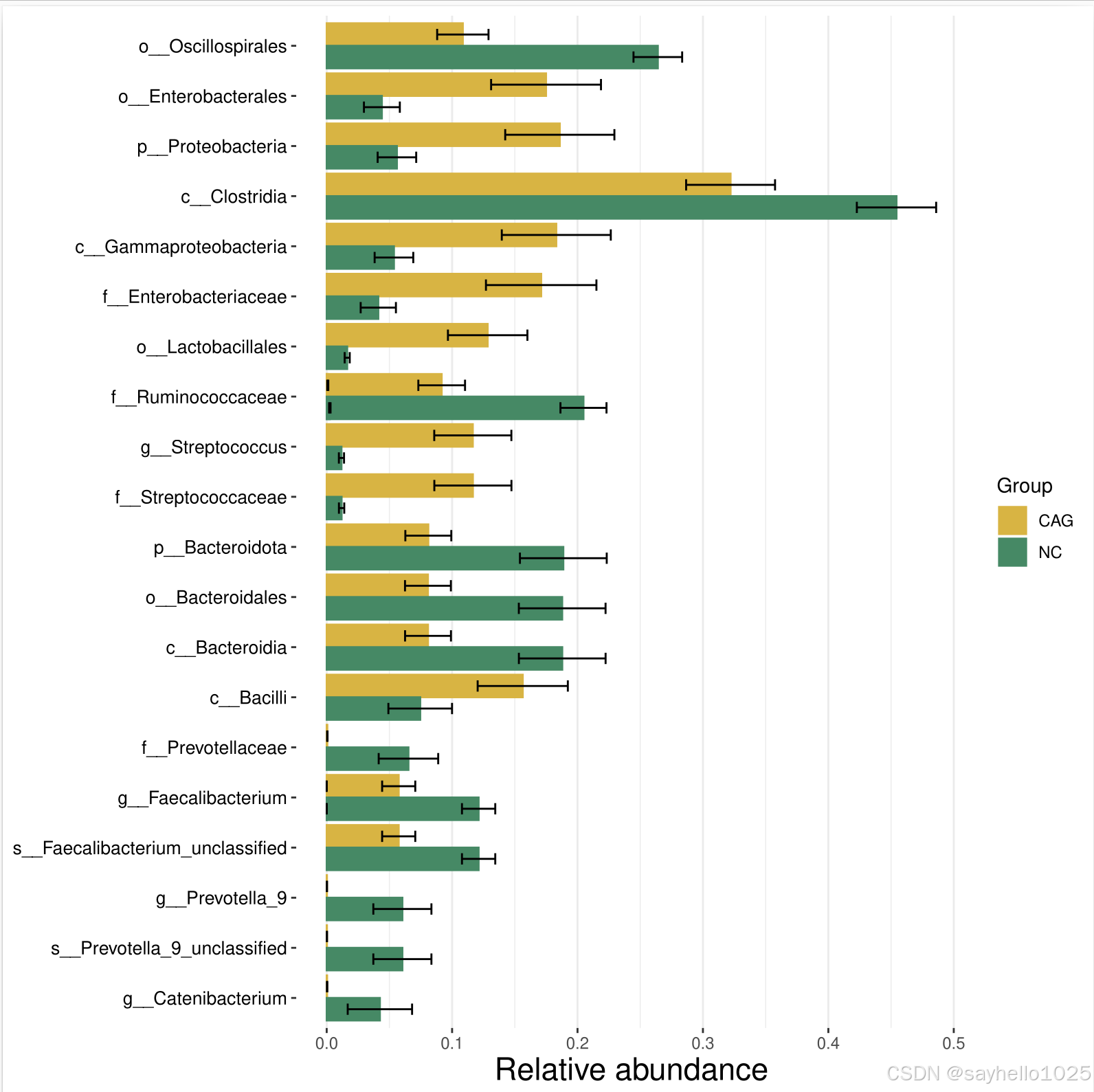
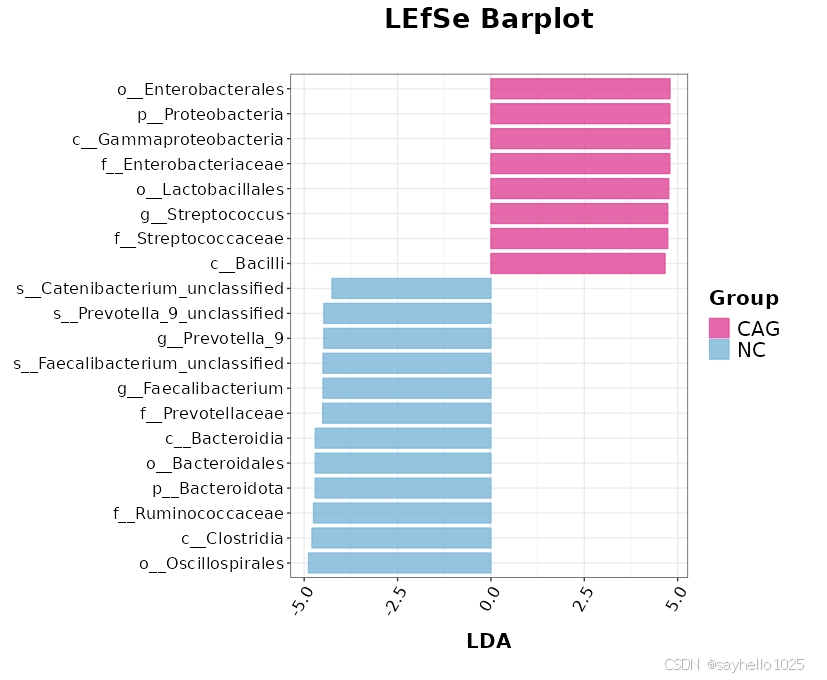
这张 LEfSe(Linear Discriminant Analysis Effect Size)柱状图 展示了 CAG(慢性萎缩性胃炎组) 和 NC(正常对照组) 在微生物分类单元(从目级 o__ 到属级 g__)的显著差异菌群,并通过 LDA(线性判别分析) 评估不同菌群在两组之间的富集情况。
X 轴(LDA 分数):数值越大,表明该菌群在相应的组中富集程度越高。
颜色区分:
粉色(CAG 组):这些菌群在 CAG 组 显著富集,可能与 CAG 的发病机制有关。
蓝色(NC 组):这些菌群在 NC 组 显著富集,可能与健康胃肠道的稳态维持有关。
- CAG 组(粉色)富集菌群的意义
在 CAG 组中富集的菌群主要是 变形菌门(Proteobacteria)和乳酸菌类(Lactobacillales, Streptococcus):
(1) o__Enterobacterales(肠杆菌目)、p__Proteobacteria(变形菌门)
CAG 组显著增加,表明促炎菌群扩增,可能导致或加重胃肠道炎症。
变形菌门(Proteobacteria)富集通常与胃肠道疾病、炎症和代谢紊乱相关。
Enterobacterales 属于 革兰氏阴性菌,可产生 脂多糖(LPS,内毒素),激活炎症通路(如 NF-κB),可能加剧胃黏膜损伤。
(2) c__Gammaproteobacteria(γ-变形菌纲)、f__Enterobacteriaceae(肠杆菌科)
这类细菌是肠道病原菌的常见成员,可能与肠道屏障受损和炎症有关。
CAG 组的胃黏膜可能更容易受到这些菌群的侵袭,进一步加重胃部损伤。
(3) o__Lactobacillales(乳酸菌目)、g__Streptococcus(链球菌属)、f__Streptococcaceae(链球菌科)
CAG 组乳酸菌(如 Streptococcus)显著增加,可能由于 胃酸减少,导致本来主要在口腔定植的链球菌扩增至胃部和肠道。
链球菌属(Streptococcus)偏向糖酵解代谢,生成乳酸,可能影响肠道 pH 值,进一步促进病原菌的生长。
可能与幽门螺杆菌(H. pylori)形成共生关系,促进 CAG 的发展。
(4) c__Bacilli(芽孢杆菌纲)
一些 Bacilli 可能耐受酸性环境,并在胃部疾病中发挥作用。
在胃酸减少的 CAG 组中,这类菌可能作为继发性菌群扩增。 - NC 组(蓝色)富集菌群的意义
在 NC 组中富集的菌群主要是 厚壁菌门(Firmicutes)、拟杆菌门(Bacteroidota)、瘤胃球菌科(Ruminococcaceae),这些菌群通常与健康胃肠道功能、抗炎和短链脂肪酸(SCFA)代谢相关。
(1) o__Oscillospirales(振荡单胞菌目)、c__Clostridia(梭菌纲)
这些菌群通常富含 丁酸生成菌(如 Faecalibacterium),有助于维持肠道稳态和能量代谢。
Oscillospirales 具有抗炎特性,并促进脂肪酸代谢,其在 NC 组富集可能表明其在健康状态下的重要作用。
(2) f__Ruminococcaceae(瘤胃球菌科)
该科细菌通常在健康个体中较丰富,参与膳食纤维降解,并促进短链脂肪酸(SCFA)合成。
CAG 组减少可能表明 SCFA 生成降低,影响胃肠道代谢平衡。
(3) p__Bacteroidota(拟杆菌门)、o__Bacteroidales(拟杆菌目)、c__Bacteroidia(拟杆菌纲)
拟杆菌门(Bacteroidota) 是健康肠道菌群的重要组成部分,能够:
参与 胆汁酸代谢,调节宿主能量代谢。
促进 脂肪酸生物合成,有助于维持胃肠道健康。
在 CAG 组减少,可能意味着 脂肪酸代谢紊乱、胆汁酸代谢异常和 SCFA 生成减少。
(4) g__Faecalibacterium(粪杆菌属)、s__Faecalibacterium_unclassified
Faecalibacterium 是重要的抗炎菌,可产生丁酸(Butyrate),维持肠道屏障功能。
在 CAG 组显著减少,可能加剧胃肠道炎症,并影响脂肪酸代谢。
(5) g__Prevotella_9、f__Prevotellaceae(普雷沃菌科)
Prevotella 属细菌通常与健康饮食和 SCFA 代谢相关。
在 NC 组富集,可能有助于维持胃肠道稳态
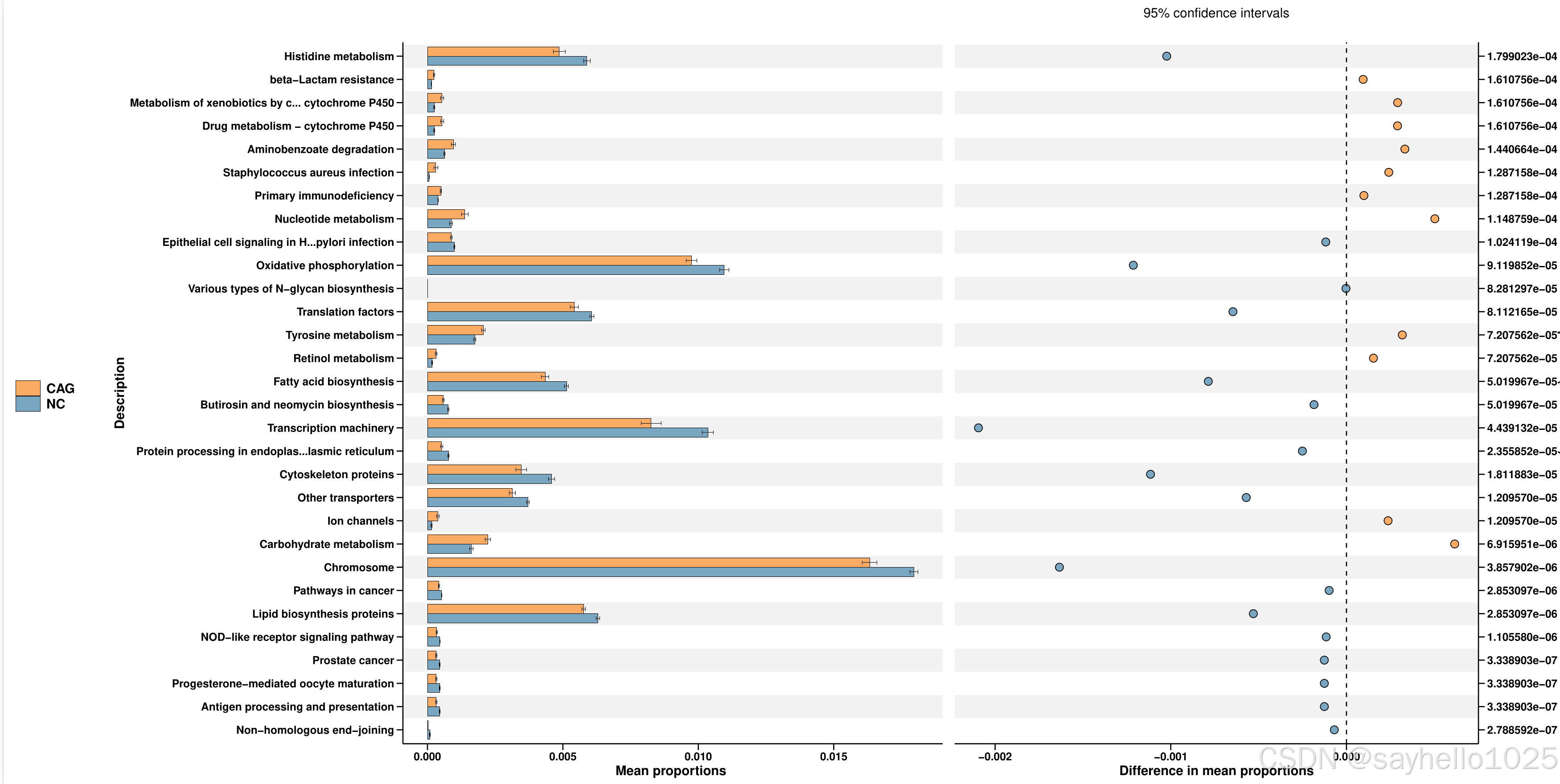
该图表显示了 CAG(慢性萎缩性胃炎组) 与 NC(正常对照组) 在微生物功能预测通路上的差异。主要的解析维度包括:
左侧柱状图(Mean proportions):
代表每个功能通路在 CAG(橙色)和 NC(蓝色) 组中的相对丰度。
误差棒代表置信区间(95% CI)。
右侧散点图(Difference in mean proportions):
代表两组之间功能通路的差异(CAG - NC)。
正值(右侧):CAG 组显著高于 NC 组。
负值(左侧):CAG 组显著低于 NC 组。
- 主要发现
(1) CAG 组显著升高的功能通路(右侧散点图,橙色点)
与病原菌和炎症相关的通路增加:
Histidine metabolism(组氨酸代谢)
beta-Lactam resistance(β-内酰胺耐药性)
Metabolism of xenobiotics by cytochrome P450(细胞色素 P450 代谢外源物质)
Drug metabolism - cytochrome P450(细胞色素 P450 药物代谢)
Staphylococcus aureus infection(金黄色葡萄球菌感染)
Primary immunodeficiency(原发性免疫缺陷)
Epithelial cell signaling in H. pylori infection(幽门螺杆菌感染中的上皮细胞信号通路)
✅ 解读:CAG 组病原菌信号通路增强
H. pylori 感染相关通路显著增加,可能表明 CAG 组的微生物群落中 幽门螺杆菌或其他病原菌的活性增强,导致胃部免疫信号异常。
β-内酰胺耐药性增加,提示 CAG 组可能存在更多耐药菌群,这在慢性炎症环境下可能与长期抗生素暴露相关。
P450 相关通路升高,提示 细菌可能通过药物代谢或毒性分子代谢增强 适应炎症环境。
(2) CAG 组显著降低的功能通路(左侧散点图,蓝色点)
与能量代谢、免疫调控和脂肪酸代谢相关的通路减少:
Oxidative phosphorylation(氧化磷酸化)
Translation factors(翻译因子)
Tyrosine metabolism(酪氨酸代谢)
Retinol metabolism(视黄醇代谢)
Fatty acid biosynthesis(脂肪酸生物合成)
Butirosin and neomycin biosynthesis(布替霉素和新霉素生物合成)
Lipid biosynthesis proteins(脂质生物合成蛋白)
NOD-like receptor signaling pathway(NOD 样受体信号通路,参与炎症应答)
Antigen processing and presentation(抗原加工和呈递)
✅ 解读:CAG 组能量代谢下降
脂肪酸生物合成减少,支持之前的分析,即 CAG 组的微生物代谢向糖代谢偏移,而非脂肪酸代谢,可能导致胃肠道能量供应不足。
氧化磷酸化(Oxidative phosphorylation)下降,表明 CAG 组微生物的能量代谢能力降低,可能影响宿主代谢稳态。
抗原加工和呈递下降,提示 宿主免疫识别能力可能受损,可能影响抗感染能力。
2. 结合微生物学解释
CAG 组的微生物群落更倾向于致病菌(Proteobacteria, Enterobacterales, Streptococcus)
这些菌群通常具有较强的耐药性和病原相关基因,这可能解释 β-内酰胺耐药性增强。
H. pylori 相关信号通路增加,可能提示幽门螺杆菌在 CAG 组的高丰度。
NC 组的微生物群落富含短链脂肪酸(SCFA)生成菌(Bacteroidota, Faecalibacterium, Oscillospirales)
这些菌群主要参与 脂肪酸生物合成和能量代谢,其减少可能导致 CAG 组脂肪酸生物合成下降。


















 6995
6995

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









