









Iterative Transfer Learning with Neural Network for Clustering and CellType Classification in Single-cell RNA-seq Analysis ——20210301

Background
单细胞RNA测序(scRNA-seq)分析的一个重要步骤是通过聚类来确定细胞群或类型。细胞类型的知识可以揭示细胞在组织、发育阶段和生物体中的异质性,并提高我们对细胞和基因功能在健康和疾病中的理解。但其高维性和遗传的高水平技术噪声是细胞类型识别的主要障碍。流行的scRNA-seq聚类方法,如Louvain、SIMLR和SC3,对于细胞类型密切相关或测序深度较低的数据可能表现不佳。虽然去噪方法可以提供更准确的基因表达估计和帮助聚类,这些方法是无监督的,不能利用细胞类型特异性基因表达信息。由于大量注释良好的scRNA-seq数据集已经可用,许多最先进的方法开始利用这些数据集中的信息来帮助在新数据中识别细胞类型。由于源数据和目标数据提供了不同数量的细胞类型特异性基因表达信息,研究者希望使用数据驱动的方法来确定每个数据类型在分析中的贡献。
转移学习是一种机器学习方法,它专注于存储在解决一个问题时获得的知识,并将其应用到另一个不同但相关的问题上。借用这个想法,研究者开发了一种监督机器学习方法ItClust,它利用从源数据中学习到的细胞类型特异性基因表达信息,帮助在新生成的目标数据上对细胞类型进行聚类和分类,实现自动确定目标数据集中的集群数量,分离源数据中缺少的细胞类型。
Methods
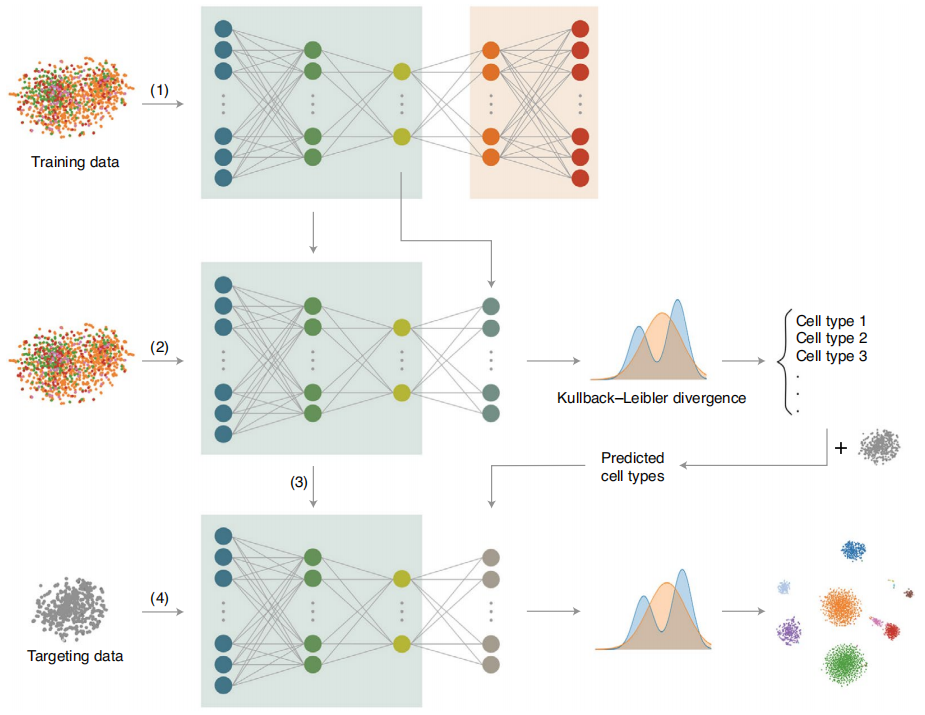
Fig. 1 Overview of the ItClust framework.
首先,ItClust需要两个输入数据集:一个源数据集,其中包括带有良好注释的细胞类型标签的细胞;一个目标数据集,其中包括需要聚集和注释的细胞(图1显示了ItClust算法的概述)。ItClust从构建源网络开始,从源数据中提取细胞类型特异性基因表达特征。经过参数优化后,该源网络可以初始化第二个源网络,即目标网络,由源网络估计参数。然后使用目标数据中的细胞进一步训练初始化的目标网络,以微调参数,以便捕获目标数据中的细胞类型特异性基因表达特征。一旦完成微调,目标网络将返回目标数据中的集群细胞。将ItClust与现有的方法进行比较,结果表明,ItClust在聚类和细胞类型分类方面始终优于现有的方法。
Motivation for the use of transfer learning
随着scRNA-seq研究的规模不断扩大,产生了越来越多的数据。许多研究是多年的项目,随着时间的推移,不断产生新的数据。每次生成新数据时重复聚类和细胞类型分类分析是非常耗时的。为了简化分析,在利用从以前的数据分析中获得的细胞类型信息的同时,对新生成的细胞进行聚类和注释是很有吸引力的。带有监督前训练的迁移学习非常适合于此目的,因为先前的聚类和注释的细胞可以用于训练监督模型,新生成的细胞可以受益于利用从先前模型学习到的基因表达特征。
Information extraction from source data using the source network.
ItClust需要两个输入数据集:一个源数据集包括带有良好注释的细胞群类型标签的细胞群,另一个目标数据集包括需要聚类和注释的细胞群。ItClust首先从目标数据中选择h个高变异基因。然后在源数据中找到p个重叠的基因**(p≤h),提取源数据中所有n个细胞对应的基因表达矩阵。设X ∈ \in ∈ R n×p** 为提取的正态化单细胞基因表达矩阵,用于训练源网络,其中行对应细胞,列对应基因。当scRNA-seq是高维数据,则不直接用原始数据训练,而是使用非线性映射函数fθ:X → \rightarrow →Z 对数据进行降维,其中θ表示嵌入参数,Z ∈ \in ∈ R n×d 是潜在的特征空间的维度d( d ≪ p d \ll p d≪p)。为了参数化fθ,我们使用一个堆叠自动编码器,一层一层地初始化它,每一层都是经过训练的去噪的自动编码器,以重建随机损坏后的前一层的输出。除了瓶颈层Bottleneck Layer 和最后解码器层last decoder layer tan h作为激活函数外,其余的使用ReLU作为激活函数。在以最小二乘损耗最小的方式训练每一层之后,我们将所有编码器层和所有解码器层按反向分层训练顺序连接起来,形成一个多层自动编码器,中间有一个瓶颈层,并对其进行微调,使重构损耗最小。然后我们丢弃所有解码器层,并使用编码器层作为原始数据和降维特征空间之间的初始映射。
下一步添加一个集群层Clustering Layer到编码器网络。由于对源数据集进行标记,并且真正的细胞群的数目为k是已知的。对于每个簇j (j=1,…,k),簇心μj是根据每个细胞类型的细胞在嵌入层中的平均特征来确定的。这一步为每个细胞类型分配一个初始簇,并确保在随后的网络优化过程中,只有细胞类型特定的基因表达特征被捕获。
Transferring cell type information in the source data to the target network
将源数据中的细胞类型信息传输到目标网络。设X’ ∈ \in ∈Rm × \times ×p为m个细胞的目标数据集的正态化基因计数矩阵,其中p是源数据中存在的目标数据中高度可变基因的子集。我们用与源网络相同的结构建立一个新的网络。我们不是随机初始化目标网络,而是将从源网络学习到的权值作为初始值转移到目标网络,除了最终的聚类层。这一步确保了新网络可以像在源网络中那样将目标数据映射到相同的特征空间Rd,也就是X’ → \rightarrow →Z’(Z’ ∈ \in ∈Rm × \times ×d)。下一步,我们在特征空间Z上应用迭代聚类算法。细胞类型标签为目标数据集是未知,为了获得目标数据集中每个细胞的聚类和**聚类中心(cluster centroids)**的数量,我们使用源网络预测它的聚类类型,并使用预测数量的聚类和在每个预测细胞功能为重心的平均特征来作为初始化聚类层的聚类中心。目标数据的聚类数量取决于源数据中存在的细胞群类型的数量。具体来说,ItClust将生成与源数据中细胞类型数量相同的聚类中心数量。这些中心用于聚类目标数据中的细胞。如果没有细胞被分配到中心,它将被剔除。
Fine-tuning of the target network
目标网络的微调。除了合并来自源数据的信息之外,还需要合并仅存在于目标数据中的唯一信息。为此,我们反复微调目标网络,使其更适应目标数据。在微调期间,对于同时出现在源数据和目标数据中的细胞类型,它们的聚类中心将主要由源数据驱动,并且在迭代过程中它们将轻微移动以适应目标数据。对于仅出现在源数据中的细胞类型,将没有(或只有少数)目标数据中的细胞分配给它们的中心,我们称这些中心为自由中心。对于目标数据特有的细胞类型,尽管没有初始中心分配给它们,但这些自由中心可以移动,并在迭代过程中用于聚集目标数据中那些看不见的细胞类型。由于这个原因,源数据中唯一的细胞类型可以为ItClust提供更大的灵活性,并帮助对目标数据中唯一的细胞类型进行聚类。因此,ItClust更倾向于使用具有综合cell类型(comprehensive cell types)的源数据集,这样的数据集可以通过将多个标记良好的数据集组合在一起来获得。
Parameter optimization for the source and target networks
源和目标网络参数优化。使用深度嵌入聚类算法对源网络和目标网络进行优化。首先,我们定义了从一个细胞到聚类质心的距离度量。根据van der Maaten和Hinton31,我们用学生st分布作为核来测量细胞i的嵌入点zi=fθ(xi) Z与聚类j的质心μj之间的距离:qij 1 þ zi μj 2=α 1 Pj0 1 þ zi μj0 2=α 1;其中α是学生st分布的自由度,在我们的实现中设为1。距离qij也可以解释为将细胞i分配到聚类j的概率。接下来,我们通过基于qij定义辅助目标分布P来迭代细化聚类。P的选择对产品的性能有重要的影响。我们定义辅助目标分布为:pij q2 ij=Pni 1 qij Pkj 1 q2 ij=Pni 1 qij;该方法提高了赋值为高置信度的细胞的权重,并将每个质心对整体损失函数的贡献归一化,以防止大型聚类扭曲隐藏特征空间。现在我们已经软任务qij和辅助分布j,我们可以将目标函数定义为Kullback Leibler(吉隆坡)发散损失:L KLðÞPjjQ Xni Xkj 1 pijlog j qij:网络参数和集群重心同时优化通过最小化L用随机梯度下降势头。得到L对zi和μj的梯度:L zi α þ 1 α Xkj 1 1 þ zi μj 2 α 0B@ 1CA 1 pij qij zi μj;j Lμαþ1αXni 1 1þ子μj 2α0 b@ ca 1 j qij ziμj:让t是当前迭代梯度L =子我用于标准的反向传播网络s参数计算梯度L =θ我和L =μj,用于更新聚类质心完每一批大小b(例如,b = 256)由μtþ1 j Lμμtj tjμtj bxbi 1 1þ子μtj 2α0 b@ ca 1 j qij ziμtj:当满足这两个条件之一时,算法将停止:(1)对目标数据训练的迭代次数超过2000次,这是我们设定的最大迭代次数;(2)当前迭代和上一次迭代中具有不同预测细胞类型标签的细胞百分比小于0.1%,这是ItClust默认的容忍水平。
Architecture of the neural network in ItClust.
ItClust中的神经网络结构。每一层的层数和节点数取决于源数据中的样本大小。较大的源数据通常包含更多关于细胞类型的信息,并且通常需要更大的网络来存储这些信息。我们建议在编码器中使用不同数量的隐藏层和不同数量的节点。补充表2给出了ItClust中隐藏层和节点的默认数量。在补充表4中,我们显示了层数和每层节点的数量对ItClust的性能影响很小。
Cell type assignment
细胞类型的任务。除了聚类之外,我们还希望利用源数据中的细胞类型信息来帮助在目标数据中聚类细胞类型。与预测每个单个细胞的细胞类型的大多数受监督的细胞类型分类方法不同,ItClust为每个聚类提供了一个置信度评分confidence score,以帮助为该聚类中的所有细胞进行细胞类型注释。由于源数据集被很好地标记,在微调之前,我们模型中的每个聚类中心表示源数据中存在的已知细胞类型。例如,假设细胞群i用于在进行微调之前对源数据中的细胞类型i进行集群。我们首先使用预微调模型为目标数据中的细胞分配集群。让Ai;在我表示目标数据中分配给簇I的细胞集之前。由于预先微调的模型仅在源数据(Ai中的细胞)上训练;在我拥有类似源数据中的细胞类型I的基因表达模式之前。迭代调整期间,我作为集群的重心不断更新其位置,目标数据可能被添加到一些细胞和其他细胞可能从集群我删除。调整完成后,我们得到了另一组细胞,人工智能,我后,由我分配给集群的所有目标细胞。如果集群的质心我仍用于集群细胞类型在目标数据,I之前也应该出现在Ai中;I之后。基于这个想法,信心得分为每个集群可以计算的原始细胞比例最终集群:si # Ai;前\ Ai; # Ai;后:这种信心分数衡量相信细胞在特定集群目标数据是相似的细胞类型,存在于源数据。
Data preprocessing and quality control
数据预处理和质量控制。ItClust可以处理不同格式的数据,包括UMI count、FPKM、TPM等。所有数据都遵循相同的预处理过程。
-
首先,对于源数据和目标数据,如果非零表达基因的数量小于100个,就会过滤掉一个细胞。
-
我们进一步去除MT和ERCC基因以及在少于10个细胞中表达的基因;然后对基因表达式值进行规范化。
- 在第一步中,细胞水平正态化,即每个细胞中的每个基因表达除以细胞中的总基因表达水平,再乘以10,000,然后转化为自然对数刻度。
- 在第二步中,执行基因水平归一化,通过减去所有细胞的平均值,再除以给定基因所有细胞的标准偏差,对每个基因的细胞水平归一化值进行标准化。使用Scanpy package32中的filter_genes_dispersion函数选择高度可变的基因(https://github.com/theislab/scanpy)。高变异基因的选择仅基于目标数据集。通过这样做,我们可以确保所有选择的基因都有信息来区分目标数据中的细胞。
-
接下来,我们发现目标数据中高度可变的基因与源数据中存在的基因之间的重叠,然后我们从源数据中提取这些重叠基因的表达模式。
Downsampling experiments on UMI counts
UMI计数的下采样实验。我们只对带有UMI计数的数据集进行了下采样实验(Downsampling Experiments)。为了从原始的scRNA-seq数据中生成下采样数据集,我们从原始数据集中选择了高质量的细胞和高表达基因。以c细胞中g基因的表达量作为的真实表达量** λ \lambda λgc**。我们利用平均参数** τ \tau τc λ \lambda λgc的泊松分布(Poisson Distribution)生成下采样数据集,其中 τ \tau τc为细胞比效率;这确保下采样的数据集和原始数据集在均值表达式和零项的百分比上是相似的。为了模拟各细胞效率的变化,我们的样本 τ \tau τc如下: τ \tau τc ≈ \approx ≈gamma的效率为75% (10,0.075); τ \tau τc ≈ \approx ≈gamma为50% (10,0.0.05); τ \tau τc ≈ \approx ≈gamma**(10,0.025)为25%; τ \tau τc ≈ \approx ≈gamma(10,0.01)为10%。
Results
Overview of the method and evaluation
为了表明从标记良好的源数据中整合细胞类型特异性基因表达信息有助于目标数据的聚类,研究者将ItClust与无监督聚类算法进行比较。研究者分析了四个公开的人类胰岛数据集。首先对四个目标数据集分别进行分析,然后将它们组合在一起形成一个数据集,以测试ItClust在目标数据中存在批处理效应时是否稳健。采用调整后的Rand指数(ARI)作为聚类准确性的评价指标。图2a显示,随着分辨率参数的变化,Louvain、DESC和SAVER-X的ARIs在所有四个独立目标数据集上都有显著变化。相比之下,ItClust不需要分辨率参数的规范,即使与Louvain、DESC或SAVER-X使用的性能最好的分辨率相比,它也总是具有最高或接近最高的ARI,且ItClust保持聚类精度高和强劲的一批效果在目标数据(图2b)。研究中发现ItClust等监督方法不需要在目标数据中包含批处理信息,清楚地显示了这些小细胞类型的单独簇(图2c)。上述结果表明,ItClust通过对标记良好的细胞的源数据进行训练,提取出每一种细胞类型的基因表达特征,从而避免捕获批量效应的信息,使其对稀有细胞的检测更加敏感。
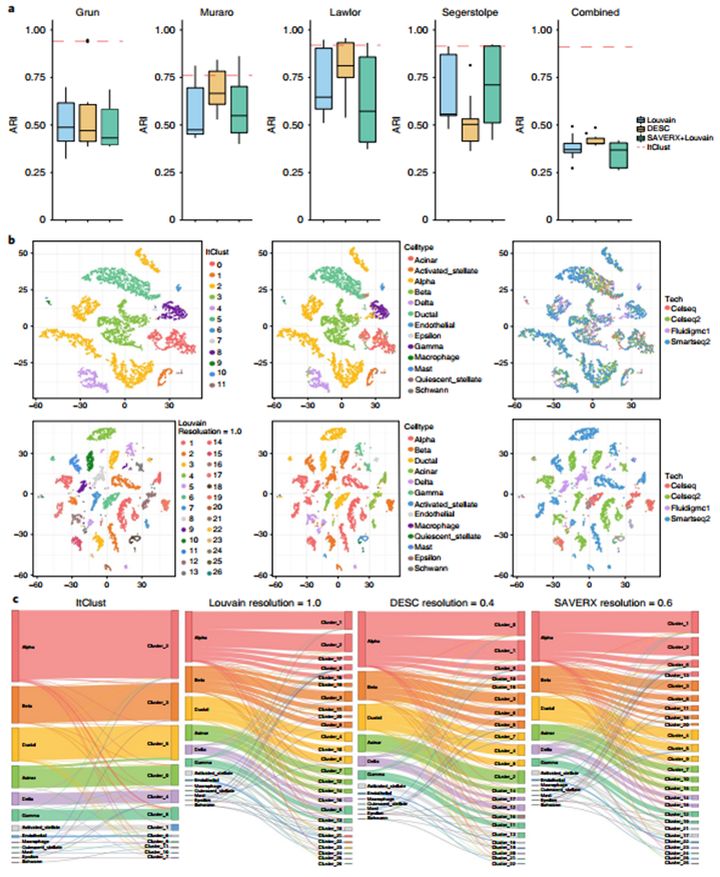
Fig. 2 Comparison of ItClust with unsupervised methods on human pancreatic islet datasets.
Comparison with unsupervised clustering methods
然后,研究者比较了ItClust与半监督和监督细胞类型分类方法。除了集群之外,ItClust还为每个集群提供一个置信度评分,它表示目标数据中的集群与源数据中的注释细胞类型的相似性程度。可以根据源数据中相应的注释为具有高置信度分数的集群分配细胞类型名称,而具有低置信度分数的集群可能表示源数据中不存在的细胞类型。首先,考虑了源数据和目标数据来自同一物种的情况。使用先前分析的四组人类胰岛数据集作为目标数据,Baron等人的数据作为源数据。当分别考虑四个目标数据集时,ItClust总体上取得了最好的性能,产生了最高或接近最高的分类精度(图3a)。当四个目标数据集合并为单个目标数据集时,ItClust仍然达到了0.95的高精度,每个聚类对应一种细胞类型(图3b),说明它对目标数据的批处理效应具有稳健性。ItClust通过直接利用标记良好的源数据中的细胞类型信息来获取目标数据的初始集群中心,从而避免了这个问题。
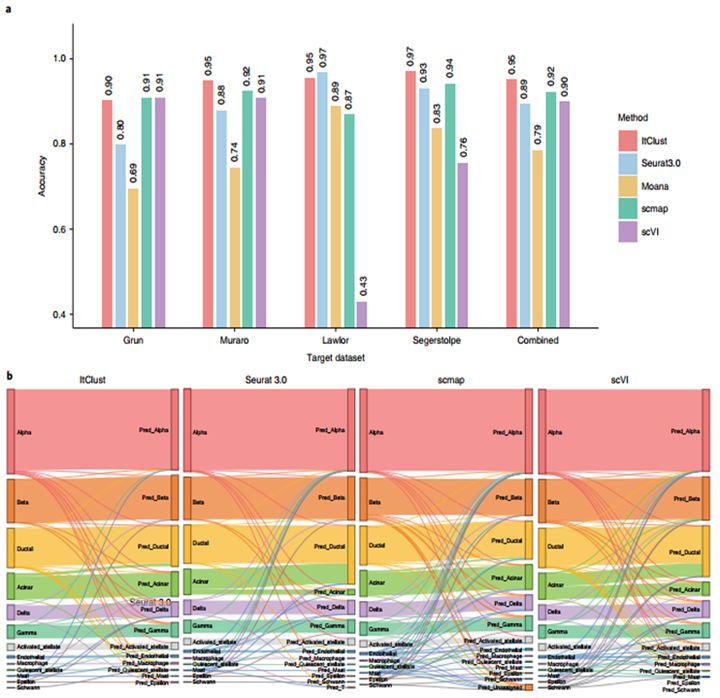
Fig. 3 Comparisons of ItClust with semi-supervised and supervised methods on human pancreatic islet datasets when source and target data are from the same species
**Comparison with semi-supervised and supervised cell type classification methods. **
接下来,将从一个物种学到的细胞类型知识转移到在不同物种中生成的目标数据集。选择人的肾脏信息,使用DESC对人类数据中的细胞进行聚类。如图4a所示,ItClust获得了最高的细胞类型分类精度(0.87),远高于表现第二好的方法Seurat 3.0(0.69)。其中Seurat 3.0错分了超过一半的巨噬细胞为成纤维细胞,而ItClust正确标记了94.6%的巨噬细胞(图4b)。为了进一步验证这些结果,进一步选择了巨噬细胞和成纤维细胞的标记基因,分别生成了真实细胞类型和ItClust和Seurat 3.0预测的细胞类型的基因表达点图(图4c)。在此基础上,研究者在胰腺中进行了跨物种转移分析。尽管目标数据很简单,但跨物种转移却很困难,因为转移仅存在于小鼠而不存在于人类的基因表达标记噪音的风险。如图4d所示,ItClust仍然取得了最高的分类准确率(0.89),远远高于第二好的方法scVI (0.27);图4e所示结果进一步证实了这一点。与其他方法相比,ItClust的一个关键优势是迭代微调步骤,它能够提取区分目标数据中的细胞类型的有用信息,这一步区别于其他监督细胞类型分类方法。
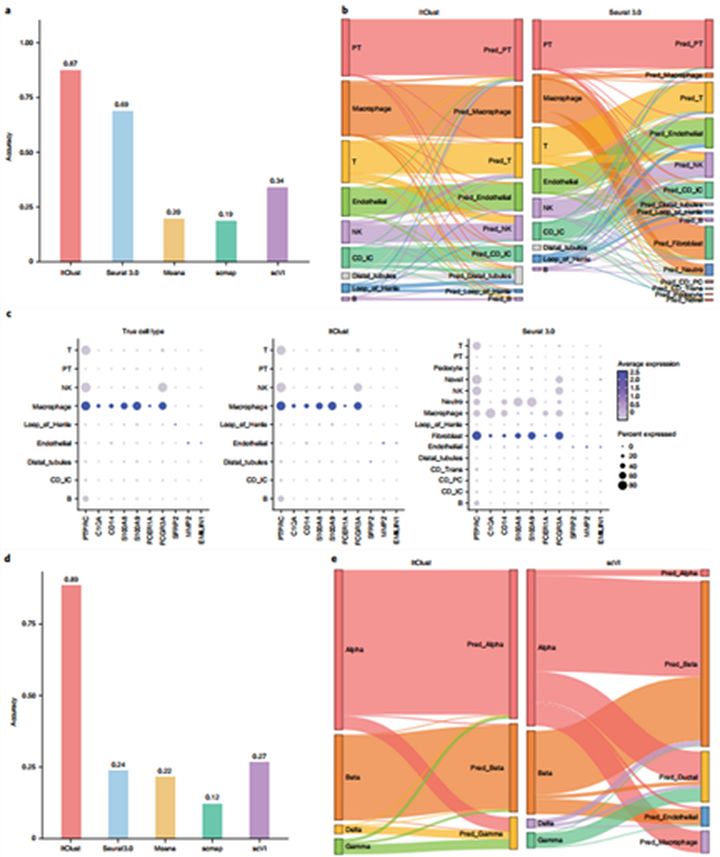
Fig. 4 Comparisons of ItClust with semi-supervised and supervised methods on mouse and human kidney datasets when source and target data are from different species
在许多研究中,目标数据可能包含源数据中没有的细胞类型。为此,研究者测试了目标数据中的独特细胞类型如何影响不同方法的性能。考虑到目标数据中有罕见的细胞类型,但源数据中没有。通过对数据进行剔除后分析,发现ItClust仍然达到了0.96的高精度。其他方法的准确率也较高。接下来对于源数据中排除的四种主要的细胞类型。图5b显示,尽管源数据中没有这四种细胞类型,ItClust仍然达到了0.92的高精度,并且能够正确地分离它们。相比之下,Seurat 3.0的准确性大幅下降到0.25,因为它将四种缺失的细胞类型中的大多数分配为导管细胞(图5c)。ItClust可以分离这些看不见的细胞类型作为我们的模型在微调步骤捕获的信息,这些独特的细胞类型在目标数据更新网络参数。通过反复微调,根据靶数据获得的细胞类型特异性基因表达信息,将这四种细胞类型与其他细胞类型逐步分离。接下来相关细胞的分析结果表明,对于看不见的细胞类型,如果在目标数据中有足够的信息,它就能够很好地利用这些信息,从看不见的细胞类型中聚集细胞。
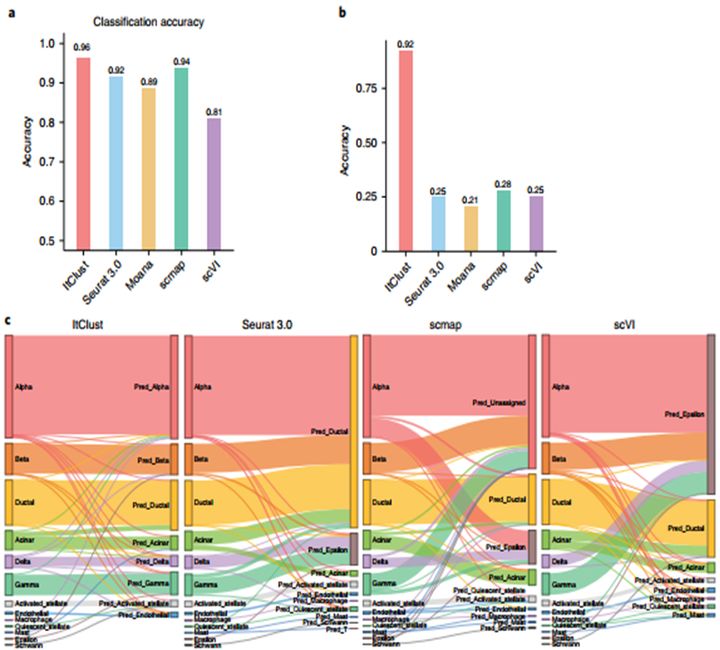
Fig. 5 Comparisons of ItClust with semi-supervised and supervised methods on human pancreatic islet datasets to evaluate the impact of missing cell types in source data.
研究者通过将两种数据集结合生成一个新的合并数据集作为源数据,ItClust的总体分类精度最高,从0.92提高到0.96。许多scRNA-seq研究存在测序深度低的问题。为了评估当源数据序列深度较低时ItClust的性能,研究者使用scRNA-seq数据集对来自4只猕猴的视网膜双极细胞进行了分析。从图5b可以看出,在效率分别为25%和10%的情况下,ItClust的微调步骤大大提高了分类精度。从源数据和目标数据中提取细胞类型特异性基因表达特征的能力使其具有灵活性。最后,对定集群中的细胞与源数据中的细胞类型相似程度进行比较。采用置信度的不同成功的将其分为不同类型的细胞。
总之,本研究提出了一个监督聚类算法ItClust,采用了迁移学习框架。研究中使用来自不同物种(小鼠、猕猴和人类)的数据集和组织(胰腺、肾脏和视网膜)对ItClust进行了广泛的测试,将ItClust与其他无监督聚类方法进行比较表明,它总是能够不需要任何超参数的调整实现较高的ARIs。与流行的监督细胞类型分类方法的比较也支持ItClust在所有评估场景中的一致高性能。此外,ItClust对于网络架构、源数据中细胞类型的误标记具有很强的稳健性,并且可以为由连续的转录表型生成的细胞保持连续的结构。

















 942
942

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









