







原文标题:Multi‑slice spatial transcriptome domain analysis with SpaDo
原文链接:Multi-slice spatial transcriptome domain analysis with SpaDo | Genome Biology | Full Text
背景
空间分辨转录组学技术通过保留空间信息彻底改变了 mRNA 表达的检测方法,从而促进了空间水平生物功能的探索[1, 2]。一般来说,空间转录组学技术可分为两类:基于高通量测序的技术[1, 3, 4]和基于荧光原位杂交(FISH)的技术[5]。基于测序的技术可对整个转录组进行高通量分析,但牺牲了空间分辨率,因为它们检测的是多个空间定义位点(称为点)上的基因表达。另一方面,基于 FISH 的技术,如 MERFISH[6]、seqFISH[7]、seqFISH+[8]、osmFISH[9]等,在实现单细胞分辨率的同时,也表现出较低的通量和有限的转录本检测能力。此外,一些基于原位测序的技术,包括 STARmap [10] 和 FISSEQ [11],也能达到单细胞分辨率,但通量较低。最近,立体测序技术(Stereo-seq)异军突起,既能达到亚细胞分辨率,又能保持高通量[12]。
尽管开发出了各种空间解析转录组技术,但空间转录组学数据的分析仍然是一项重大挑战,尤其是在空间域分析方面。空间域是指由多个细胞组成的特定空间区域,通常与组织的解剖结构和特定功能相关[13]。这些空间域可被视为包含空间信息的基本功能单元,用于下游分析。目前,已开发出多种空间域检测方法,可分为三类:(1)传统的域检测方法,包括 Seurat [14] 和 Scanpy [15] 等,这些方法没有明确考虑空间信息。这些方法通常被用作域检测的基线。(2) 基于统计模型的方法,如 BayesSpace [16] 等,是基于空间上相邻的点更有可能表现出相似的基因表达模式这一简单假设而开发的。然而,这些方法并不是专门为处理单细胞空间转录组而设计的。(3) 基于图神经网络的方法,包括 SpaGCN[17]、SEDR[18]、STAGATE[19]等,假定每个点的基因表达信息可以利用其相邻信息重建。通常情况下,每个斑点的低维嵌入(包含空间信息)会被获得。尽管如此,仍可能存在计算复杂度相对较高或可解释性有限等挑战。
此外,虽然现有方法可以检测单个组织切片内的空间域,但无法直接处理一般多个组织切片的多切片空间域分析。随着空间转录组的快速发展,可以整合多个组织切片来揭示空间细胞景观的组织切片越来越多。例如,通过整合多切片空间转录组数据,发现了心肌梗塞病变状态下具有空间依赖性的特定细胞类型,从而揭示了新的致病机制和新的治疗方案[20]。此外,多切片分析显示,三级淋巴结构(TLS)是在非淋巴组织中发现的有组织的免疫细胞群,通常与肿瘤预后的改善有关,其细胞类型组成稳定而一致[21]。因此,多切片空间转录组域分析是处理不断积累的多切片空间转录组数据的基础,尽管它带来了巨大的挑战。值得注意的是,目前已开发出几种用于切片配对的计算方法,如 PASTE [22] 和 SLAT [23]。不过,这些方法主要针对单个细胞或斑点的配准。它们不能直接用于分析多个切片的空间域,因此限制了它们的应用。
为此,我们提出了 SpaDo(多切片空间转录组域分析),用于单细胞和点分辨率的多切片空间转录组分析。具体来说,SpaDo 包含三个功能模块:多切片空间域检测、基于参考的空间域注释和多切片聚类分析。SpaDo 显示了几个关键优势,包括良好的可解释性、鲁棒性以及对噪声和批次效应的耐受性。
我们对从 7 种不同的空间转录组测序平台(包括 osmFISH、seqFISH +、STARmap、MERFISH、10 × Visium、old ST 和 Slide-seqV2 等)获得的 40 多组多切片空间转录组数据进行了全面调查,证明了 SpaDo 的优越性[4](附加文件 1:表 S1)。我们的研究结果凸显了 SpaDo 在从多切片空间转录组中获得新的生物学见解方面的巨大潜力。
方法
数据描述
SpaDo 的设计兼容所有空间转录组测序技术和平台。在本研究中,我们特别测试了 SpaDo 在 osmFISH、STARmap、seqFISH +、MERFISH、10× Visium、ST 和 Slide-seq V2 平台上的性能(附加文件 1:表 S1)。值得注意的是,DLPFC 数据集[33]包括从三个人身上取样的 12 张人类 DLPFC 切片。DLPFC 层和白质(WM)由原始研究人工标注。为了获得上述 12 张 DLPFC 切片的细胞类型丰度,我们以 DLPFC 的单细胞转录组数据[47]为参考,使用 Cell2location [26]进行了斑点去卷积。
我们研究中使用的 MERFISH 数据集[48]由三个样本组成,它们的 Animal_ ID 分别为 31、32 和 33。这些样本的 Bregma 值为 0.16,其特定行为特征被描述为 "对成体的攻击"。
我们研究中使用的 RCC 数据集[34]包括五个 RCC 切片(10 × Visium):"GSM5924041_ffpe_c_51"、"GSM5924043_frozen_a_3"、"GSM5924044_frozen_a_15"、"GSM5924046_frozen_b_1 "和 "GSM5924047_frozen_b_7"。其中,前四片含有一个或两个 TLS 区域,最后一片不含 TLS 区域作为阴性对照。我们以 RCC 的单细胞转录组数据[49](P76 和 P90)为参考,利用 Cell2location 对上述 5 张 RCC 切片进行了斑点解卷积,并将原始研究中的细胞亚型注释合并为 17 种主要细胞类型。
我们的研究中使用的人类心脏数据集[40]由 19 个切片(ST)组成,代表了怀孕头三个月中三个发育阶段的发育中人类心脏:孕后 5 周、6 周和 9 周(PCW)。我们以原始研究中的单细胞转录组数据为参考,利用 Cell2location 获得细胞类型去卷积结果。
我们研究中使用的鸡心数据集[42]由 11 张切片(10 × Visium)组成,这些切片分别取自 4、7、10 和 14 天的四腔心脏早期至晚期阶段。我们以原始研究中的单细胞转录组数据为参考,利用 Cell2location 获得细胞类型解卷积结果。
我们研究中使用的类器官数据集[41]包括 10 个切片(Slide-seq V2),分别取自发育中的人类皮质类器官 1、2 和 3 个月的切片。鉴于 Slide-seq V2 的高分辨率(每个点的直径为 10 μm,包含约 1-3 个单细胞),我们将类器官数据集作为单细胞分辨率的空间转录组数据进行分析。细胞类型标签来自原始研究。
数据预处理
我们根据生成数据所使用的空间转录组平台,采用了不同的归一化方法。对于 osmFISH、STARmap 和 MERFISH 平台生成的数据集,我们采用了各自原始研究推荐的归一化方法。这包括将每个细胞的基因计数除以每个细胞的总计数,然后进行对数变换(log(1 + 归一化计数))。对于从其他平台(包括 seqFISH +、10 × Visium、ST 和 Slide-seq V2)获得的数据集,我们使用 Seurat 软件包进行了标准归一化处理。这包括用总表达量归一化每个细胞/点的基因表达测量值,将结果乘以 10,000 的比例因子,最后应用对数变换(log(1 + 归一化计数))。
细胞类型注释
SpaDo 根据不同的分辨率,无论是单细胞还是点水平,都采用了不同的细胞类型注释策略。
对于单细胞分辨率的空间转录组学数据,在我们的研究中,我们使用了原始研究中的细胞类型标签。在无法获得这些标签的情况下,我们采用了 Seurat v4,使用默认参数选择了前 2000 个高变异基因,并使用参数 "分辨率 = 2 "获得了细胞类型注释结果。重要的是,SpaDo 对 Seurat v4 的 "分辨率 "参数的鲁棒性在我们的分析中得到了证明(图 3c)。值得注意的是,对于多切片单细胞分辨率转录组数据,Seurat 必须应用于整个多切片基因表达谱,以确保细胞类型注释结果的一致性。
另一方面,对于点分辨率空间转录组学数据,在我们的研究中,SpaDo 特别利用 Cell2location [26] 获得点注释。SpaDo 对 RCTD 和 SPOTlight 等其他斑点解卷积方法的鲁棒性也得到了证明(图 3d)。需要强调的是,对于多切片光斑分辨率转录组数据,Cell2location 应使用相同的单细胞参照物应用于整个多切片数据集,以确保光斑解卷积结果的一致性。
计算空间相邻细胞类型嵌入
SpaDo 采用了两种不同的策略,分别计算单细胞和点分辨率空间转录组数据的空间相邻细胞类型嵌入(SPACE)。对于单细胞空间转录组数据,由于 KNN 能够充分利用密度信息,因此采用 K-nearest neighbors(KNN)方法识别每个细胞的相邻细胞。然后,SpaDo 计算这些相邻细胞的细胞类型比例,得到每个细胞的 SPACE。对于点分辨率的空间转录组数据,由于点是均匀分布的,SpaDo 通过在指定半径范围内搜索,获得每个点的相邻邻居。然后,SpaDo 利用这些相邻点的解卷积结果计算细胞类型比例。
在多切片领域检测中,我们最初为每个切片生成一个 SPACE。由于所有切片的细胞类型注释一致,即共享相同的嵌入空间,因此不同切片的细胞/点的 SPACE 具有可比性。SpaDo 通过串联每个单独的 SPACE 实现了这一点,从而为多个切片获得了统一的 SPACE 表示。
空间域检测
在本研究中,空间域被定义为单张或多张切片中具有相似空间(SPACE)的细胞群或斑点。
为了检测每个切片的空间域,首先要计算 SPACE 的距离矩阵。为了测量 SPACE 的相似性,SpaDo 配备了两种距离度量,即詹森-香农发散(JSD)[50] 和曼哈顿距离。作为一种广泛使用的分布距离度量,JSD 基于两个分布之间的库尔巴克-莱伯勒发散(KL)。两个单元或点 P 和 Q 之间的 SPACE KLD 定义如下

作为一个对称、有限和平滑的版本,JSD 的定义如下:
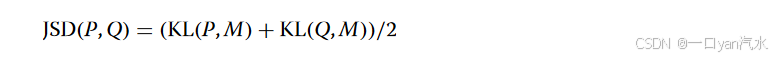
其中 M = (P + Q) / 2。JSD 值越小,表示分布之间的相似度越高,而值越大,则表示差异越大。
曼哈顿距离(MD)也是 SpaDo 软件中的一个候选项,因为它比 JSD 快得多,但牺牲了一点精确度。曼哈顿距离是各点坐标的绝对差值之和,计算公式为

接下来,SpaDo通过对距离矩阵进行层次聚类来检测空间域。使用R包自带默认参数的hclust ( )函数进行层次聚类。
对于多切片空间域检测,首先,SpaDo计算每张切片的细胞或斑点的SPACE。由于所有切片具有一致的细胞类型注释,即它们具有相同的嵌入空间,我们将每个切片的SPACE拼接在一起以计算JSD距离,然后进行层次聚类以检测空间域。最后,将每个域回溯到每个切片。
选取合适的区域编号
通过选取合适的区域编号(附加文件1 :图S1),可以得到不同分辨率的空间域。为了确定最佳空间域数,SpaDo提供了三种可选策略:( 1 )使用R包dynamicTreeCut [ 51 ]中参数为' deep Split = 2 '的cutree Dynamic ( )函数进行自动选择;( 2 )由用户根据自己的先验知识或特定需求手动设置;( 3 )可视化的层次树和UMAP聚类结果可以辅助确定最佳空间域数。后两种策略允许定制,在空间域的分析中提供了高度的灵活性和可解释性。在这项研究中,针对每个测试数据,采用了不同的方法。如果在原始研究中提供了区域标签,则将区域的数量作为空间域的数量。如果不是,则SpaDo采用第一种策略确定最优空间域数。
具有空间参考的空间域注释
SpaDo利用已注释的数据集(称为空间参考)来注释新获得的空间转录组,称为空间域查询。具体来说,该过程包括以下4个步骤:( 1 )对于空间参考中的每个空间域,通过将所有被识别为同一域的细胞/斑点的SPACE进行平均来计算质心;( 2 )计算空间查询数据集中每个单元格/点的SPACE;( 3 )计算空间查询中每个单元格/光斑的SPACE与空间基准中空间域SPACE的每个质心之间的JSD距离;( 4 )将JSD距离最小的参考中的空间域作为空间查询中对应单元格/点的标注。
多切片聚类分析
直观地说,SpaDo通过评估多个切片之间的相似性来进行多切片聚类分析。通过空间域组成计算相似度。首先,SpaDo对多张切片进行多切片空间域检测。然后,计算单个切片的空间域组成,定义为:

其中是一个向量,表示第i个切片的空间域组成,N是所有切片中检测到的域的个数。
是第i张切片中被确定为第j个结构域的细胞/斑点的数目。如果第j个区域在第i个切片中不存在,则将
设置为0。
是第i张切片中细胞/斑点的数目。
最后,SpaDo使用默认参数的R包pheatmap中的pheatmap ( )函数对所有切片的空间域组成进行层次聚类。
本研究中的参数设置
SpaDo包含了几个关键参数,包括单细胞空间转录组数据的k近邻数、点分辨率空间转录组数据的搜索半径和域数。
在本研究进行的所有测试中,k个最近邻的数量一致设置为30。论域数的选取根据试验数据的不同而不同。如果在原始研究中可用区域标签,则将区域数量作为空间域数量。在不提供区域标签的情况下,SpaDo通过R包dynamicTreeCut [ 51 ]中参数为' deep Split = 2 '的cutreeDynamic ( )函数自动选择域名。
关于搜索半径,除了人类心脏数据集[ 40 ]外,所有测试数据的默认值为" Radius = 2 "。对于人体心脏数据集[ 40 ],源自于原始ST,其中每个点的直径为100 μ m,包含约10 - 40个单细胞[ 1 ],搜索半径设置为1。这种调整是为了适应数据集的具体特征。
距离度量的敏感性
值得注意的是,SpaDo默认使用JSD计算每个点或单元格的SPACE距离。为了验证其鲁棒性,我们系统地比较了SpaDo在使用欧氏距离、曼哈顿距离、斯皮尔曼相关性、皮尔逊相关性、余弦相似性和JSD等多种距离度量时的表现。具体地,对于Spearman,Pearson和Cosine,其中结果表示在-1到1范围内的相似性,使用" 1 -相似性"获得相应的距离。该分析为SpaDo在多种距离测量方法中的稳定性提供了全面的评估。
对测序深度和dropouts的敏感性
评估了SpaDo对测序深度和dropout的敏感性,以解释空间转录组学数据中的固有噪声。具体来说,我们通过将非零表达值的10 %、30 %和50 %随机设置为零(图3g , h),人为地增加了DLPFC _ 151673和OsmFISH数据集中的dropout率。对于每个数据集,进行了n = 20的随机脱落分配。
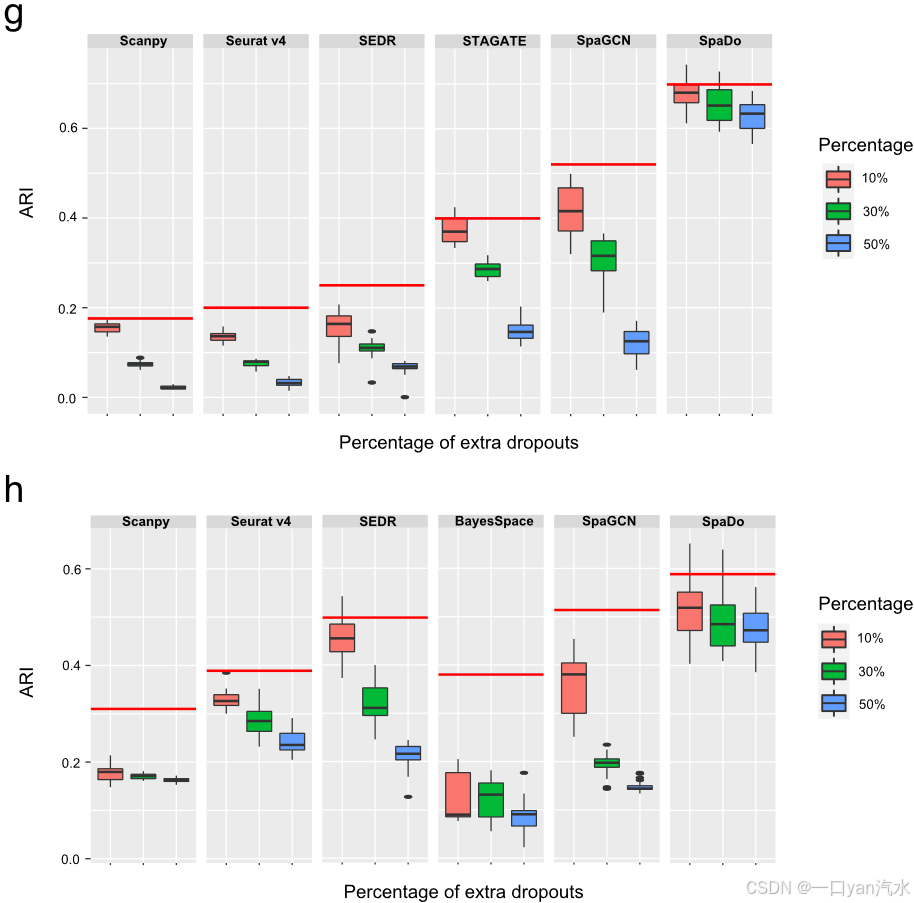
h:在DLPFC _ 151673数据集上,SpaDo在丢包率增加的情况下的表现。
额外dropout的百分比显示在情节的右边。红线表示每种方法的原始性能
SpaDo的批次效应评估
SpaDo有效地解决了批量效应,通过对4个点分辨率的DLPFC数据集( DLPFC _ 151673、DLPFC _ 151674、DLPFC _ 151675、DLPFC _ 151676)以及3个单细胞分辨率的MERFISH数据集的综合分析证明了这一点。对于光斑分辨率DLPFC数据集,我们将SpaDo嵌入策略SPACE与SEDR和SpaGCN得到的嵌入进行了比较,包括有和无和[ 38 ]。使用默认参数应用了Harmony。随后,为了评估SEDR和SpaGCN纳入和谐度后的表现,我们计算了" 1 - Pearson相关性"作为每个点嵌入之间的距离。与SpaDo不同的是,由于SEDR和SpaGCN的嵌入不是分布的,因此不适合JSD,因此在本文中不使用JSD。然后,采用与Sp Do相同的层次聚类方法对SEDR和Spa GCN进行多层域检测,指定域数为7。
对于3个MERFISH数据集,我们进行了包含SpaDo嵌入策略SPACE以及由SEDR、SpaGCN和STAGATE衍生的嵌入,有和无和的平行比较。我们遵循与上述相同的分析步骤,使用默认设置进行域数选择。
评估指标
为了评估SpaDo的性能,利用真实的空间域标签等真实信息计算了两个性能指标:调整兰德指数( ARI )和宏F1。
对于单片的空间域检测,采用ARI来评价各方法的性能:
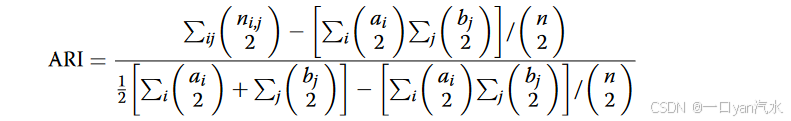
其中是被分配到第i个预测域标签的细胞数,其真实域标签为第j个标签,
和
。
对于单片的空间域检测,采用ARI来评价各方法的性能:
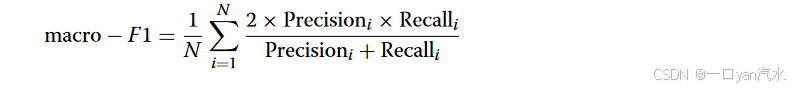
其中N表示数据集中空间域的个数。和
分别为数据集中第i个空间域的查准率和查全率。
基准测试方法
在本研究中,我们将SpaDo与Scanpy,Seurat v4,SEDR,SpaGCN,STAGATE,BayesSpace和PASTE在默认参数为(附加文件1 :表S2)的不同测试中进行了基准测试。
对于使用单细胞空间转录组数据进行空间域检测,我们使用默认参数对Scanpy,Seurat v4,SEDR,SpaGCN和STAGATE进行了基准测试。由于BayesSpace是专门针对点分辨率的空间转录组学数据而设计的,因此将其排除在这一场景之外。同原始研究设置相同的域数。
对于使用斑点水平空间转录组数据的空间域检测,我们使用默认参数对Scanpy,Seurat v4,SEDR,SpaGCN和BayesSpace进行了基准测试。STAGATE因其偶然的不稳定性和无法处理光斑分辨率数据而被排除。在原始出版物中设置相同的域数。
对于基于参考的空间域标注,我们使用默认参数对PASTE和Seurat v4进行了基准测试。
在Spa Do结合光斑反卷积方法的敏感性测试中,我们将Cell2定位与RCTD和SPOTlight进行了对比。对于Cell2location,使用默认参数训练单细胞回归模型,得到Cell2location模型,所有数据集的参数detection _ alpha = 20。具体来说,' N _ cells _ per _ location '在RCC和DLPFC数据集上设置为10,在人类和鸡心数据集上设置为20。采用默认参数进行RCTD和SPOTlight检查。
在所有对标测试中,工具均在英特尔至强微处理器E5-2696 v4 CPU ( 2.20 GHz )和精视GPU 1080 Ti的系统上执行。
结果
SpaDo概述
SpaDo是一个用于多切片空间域分析的综合计算框架,包括四个主要组件(图1):(1) 细胞类型注释,(2) 空间相邻单元类型嵌入的计算 (SPACE),(3) 基于 Jensen-Shannon divergence (JSD) 的分层聚类,以及 (4) 多切片空间域分析。
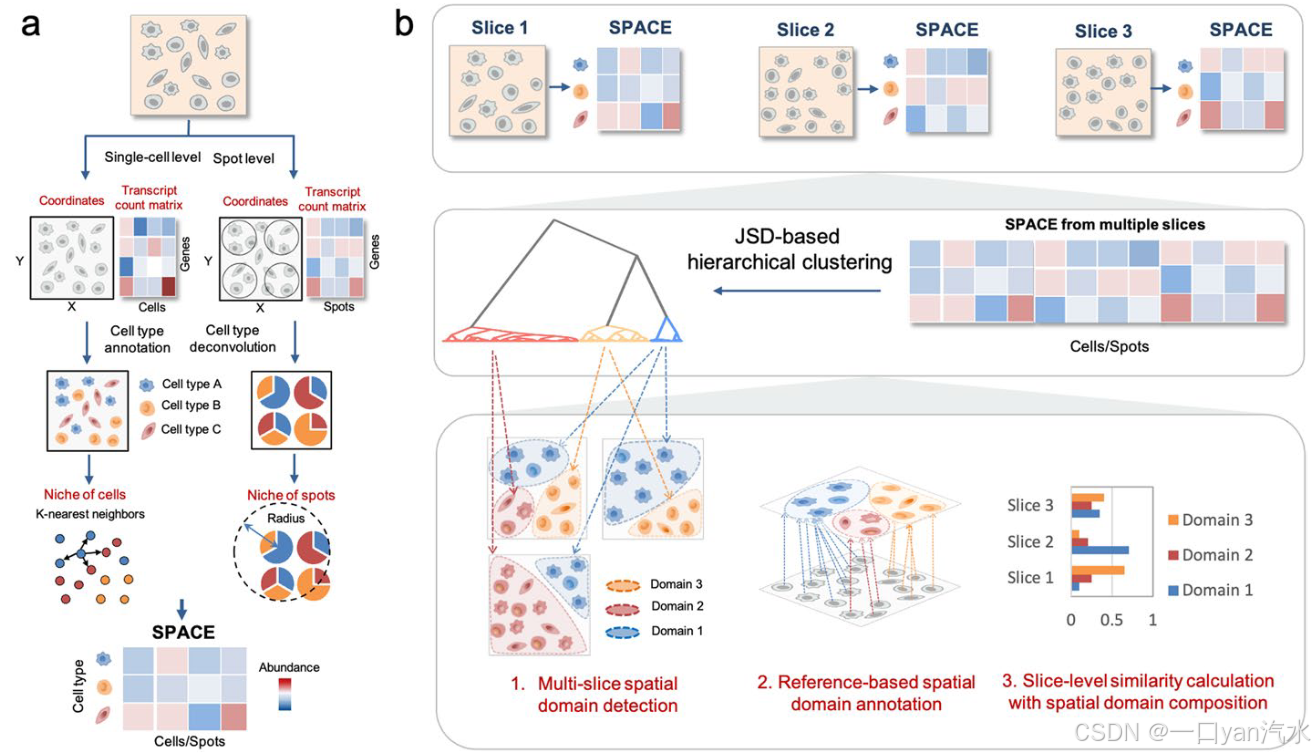
a 计算单细胞和点分辨率空间转录组数据的 SPACE。
SPACE SPatially Adjacent Cell type Embedding(空间相邻单元类型嵌入)。
坐标-转录本计数矩阵
细胞类型标注-细胞类型解卷积
细胞生态位-光斑位点
丰度
b 涉及多切片空间域分析的三个功能:多切片域检测、基于参考的空间域注释以及通过考虑空间域组成的多切片聚类分析。JSD 詹森-香农分歧
1.多切片空间域检测
2.基于参照的空间域注释
3.利用空间组合进行切片级相似性计算
SpaDo 首先要求对每个切片进行适当的细胞类型注释。根据空间转录组学数据的分辨率(单细胞或点状分辨率),会采用不同的策略(图 1a,见 "方法")。对于单细胞分辨率的空间转录组学数据,建议采用基于参考的注释方法 [24, 25] 或聚类方法,如 Seurat v4 [14]。对于点分辨率的空间转录组学数据,Cell2location [26] 被用作 SpaDo 的点去卷积方法。
其次,SpaDo 会计算空间相邻单元类型嵌入(SPACE)。如果是单细胞分辨率的空间转录组学数据,SpaDo 会通过搜索 k 个最近的邻居来确定每个细胞/点的本地生态位;如果是点分辨率的空间转录组学数据,则会搜索特定半径范围内的邻居。随后,将细胞类型注释与每个细胞/点的生态位信息融合,从而整合基因表达和空间信息,得出 SPACE。
第三,SpaDo 能够根据 Jensen-Shannon 分歧对 SPACE 进行分层聚类,从而识别多个切片中所有光点/细胞的空间域。因此,SpaDo 确定的空间域来自多个切片的组合,而不是局限于单个切片。通过回溯和映射这些检测到的空间域到每个单个切片,SpaDo 能够对多个切片的空间域进行排列,从而促进多切片空间域分析(图 1b,见 "方法")。
最后,SpaDo 支持多切片空间域分析,包括:(1) 多切片空间域检测:SpaDo 可在多个切片上检测一致的空间域;(2)基于参考的空间域注释:SpaDo 利用空间参照(即带有域注释的空间转录组数据)来注释新的测序空间转录组数据。具体来说,参考文献中的空间域标签是根据最小距离分配给查询单元格的;(3) 多切片聚类分析:SpaDo 利用空间域组成计算切片级相似性(见 "方法"),并在切片级进行聚类分析。该功能尤其适用于分析具有多个时间点或不同条件的空间转录组学数据。
与其他复杂的基于 GNN 的空间嵌入方法(如 SEDR、SpaGCN 和 STAGATE)相比,我们要特别强调 SPACE 嵌入的优势:(1)SPACE 具有很高的可解释性,非常符合空间域的生物学性质,因为空间域通常包含具有功能关系的特定细胞类型[13, 21, 27-29]。(2) SPACE 可有效解决批次效应问题,因为它是根据细胞类型的空间关系设计的,通过各种自动细胞类型注释策略[24-26, 30]和聚类方法[14, 15]确保切片间的一致性。因此,SPACE 非常适合在多个切片之间进行整合,而无需额外的批量校正。(3) SPACE 主要依赖细胞类型注释,而不是详细的空间信息。正如最近的两项研究[31, 32]所证实的那样,空间域之间的差异很明显,不需要精细的细节。这一特点使 SPACE 能够承受噪声,并对空间转录组学数据的各种变化保持稳健。
评估 SpaDo 在单片空间域检测方面的效果
我们首先对 SpaDo 和其他基于单片的空间域检测方法进行了全面评估,评估结果显示 SpaDo 可用于单细胞和点分辨率空间转录组(图 2),这为多片空间域分析奠定了基础。
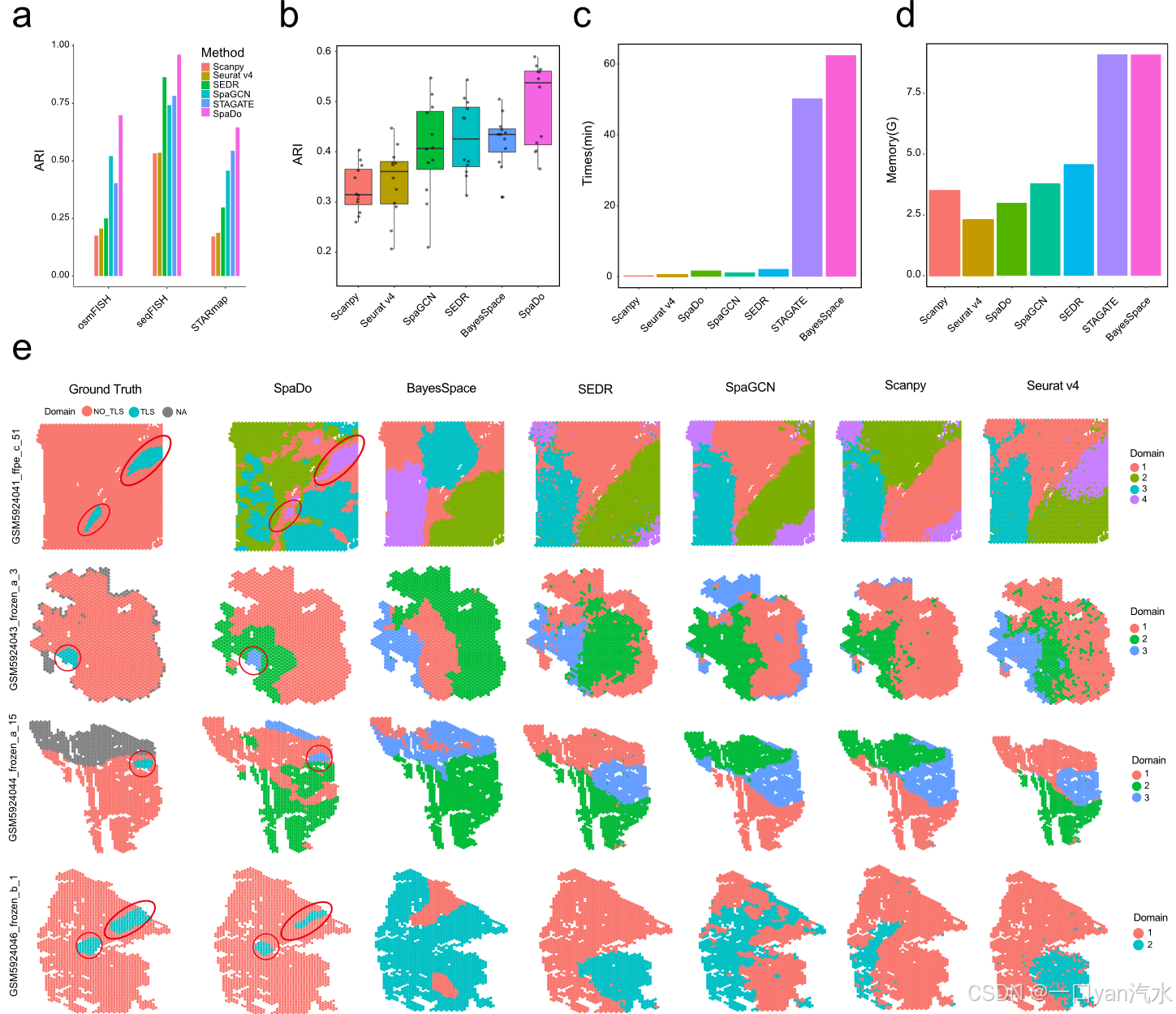
b SpaDo 和其他方法(Scanpy、Seurat v4、SpaGCN、SEDR 和 BayesSpace)在 12 个点 分辨率空间转录组数据集上的性能。
c 在 DLPFC_151673 数据(3639 个点)中测试时的单 CPU(SpaDo、Scanpy、Seurat v4、SpaDo、BayesSpace)和单 GPU(SpaGCN、SEDR、STAGATE)运行时间。
d 在 DLPFC_151673 数据中进行测试时,单 CPU(SpaDo、Scanpy、Seurat v4、SpaDo、BayesSpace)和单 GPU(SpaGCN、SEDR、STAGATE)的内存使用情况。
e SpaDo 和其他方法(BayesSpace、SEDR、SpaGCN、Scanpy 和 Seurat v4)在 4 个 RCC 空间转录组数据集上的 TLS 类域检测可视化情况
SpaDo 检测到的空间域形成了一个多分辨率的分层模式,从而可以在不同分辨率下动态地描述空间结构。为了区分不同分辨率下的空间域,我们采用了以下空间域命名方案,为特定分辨率下的每个空间域分配一个术语 ID "Domain_N1_N2",其中 N1 代表分辨率级别(即检测到的空间域数量,分辨率越高,数量越多),N2 代表特定空间域的 ID。为说明起见,我们用背外侧前额叶皮层(DLPFC)数据集[33]中的 DLPFC_151673 (10 × Visium)数据来阐明命名方案,详见附加文件 1:图 S1。此外,我们还展示了 osmFISH(附加文件 1:图 S2)和 STARmap 数据(附加文件 1:图 S3)的多分辨率空间域检测结果。
为了说明 SpaDo 在单片空间域检测方面的优越性能,我们与 Seurat v4 [14]、Scanpy [15]、SEDR [18]、SpaGCN [17]、STAGATE [19] 和 BayesSpace [16]等成熟的单片空间域检测方法进行了比较分析。我们的评估涵盖三个主要方面:(1)单细胞空间转录组学数据的空间域检测;(2)点分辨率空间转录组学数据的空间域检测;(3)类似 TLS 的域检测。
在第一个测试(图 2a)中,我们可以看到 SpaDo 在三个单细胞空间转录组数据集上的表现优于 Seurat v4、Scanpy、SEDR、SpaGCN 和 STAGATE,这三个数据集分别来自三个不同的平台:osmFISH [27]、STARmap [10] 和 seqFISH+ [8],每个数据集在域结构上都表现出不同的复杂程度(图 2a 和附加文件 1:图 S4-S6)。由于 BayesSpace 是专为点分辨率空间转录组设计的,因此我们将其排除在本次测试之外。
在第二个测试(图 2b)中,SpaDo 在 12 个点分辨率背外侧前额叶皮层(DLPFC)数据集上的表现优于 Seurat v4、Scanpy、SEDR、SpaGCN 和 BayesSpace [33]。STAGATE 因其不稳定性和偶尔出现的点分辨率数据故障而未被列入本次测试。此外,以两个 DLPFC 数据集(DLPFC_151675 和 DLPFC_151676)为例,我们展示了 SpaDo 通过整合多个切片显著提高性能的能力(附加文件 1:图 S7)。这凸显了 SpaDo 在优化结果方面的适应性和有效性,尤其是在具有挑战性的数据集场景中。
在第三项测试(图 2e)中,我们评估了四张肾细胞癌(RCC)切片[34],这些切片上有注释清楚的三级淋巴结构(TLS)区域。TLS 是一个被广泛认可的空间区域,其细胞类型组成相对一致,主要由 T 细胞和 B 细胞组成。在这项测试中,我们使用了 SpaDo 能够检测到类似 TLS 结构域的最小结构域数。这样做是为了突出 SpaDo 与其他方法相比的最高灵敏度。如图 2e 所示,SpaDo 的表现优于其他方法,它能准确检测出与人工标注标签完全一致的 TLS 样域。我们还评估了改变结构域数量的效果,发现即使选择更多的结构域,SpaDo 的表现也始终优于其他方法(附加文件 1:图 S8)。此外,SpaDo 对用户非常友好,既节省时间(图 2c),又节省内存(图 2c-d)。
SpaDo 的鲁棒性验证
由于空间转录组学数据本身存在噪声,因此分析这类数据的方法的鲁棒性至关重要。因此,我们从以下四个方面对 SpaDo 的鲁棒性进行了综合分析评估(图 3)。
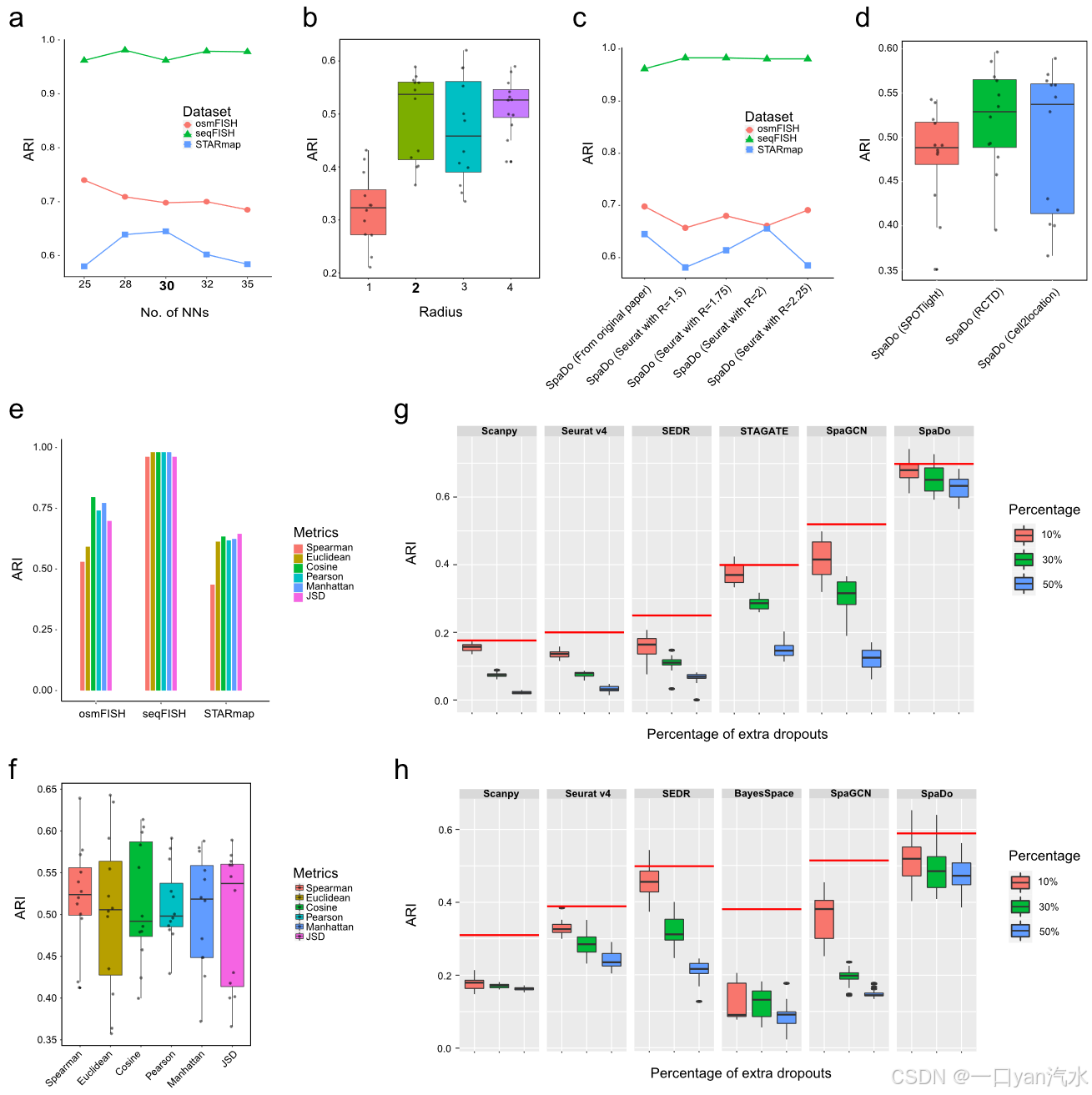
a 在三个单细胞空间转录组数据集上,不同近邻数的 SpaDo 的性能。
b 不同半径下 SpaDo 的性能。
c 在 osmFISH、seqFISH 和 STARmap 数据集上使用原始论文或 Seurat v4 中的细胞类型注释且参数 "分辨率 "值不同时,SpaDo 的性能。
d SpaDo 在使用不同点解卷积方法时的性能。
e SpaDo 在三个单细胞空间转录组数据集上使用不同距离度量时的性能。
f SpaDo 在使用 12 个 DLPFC 数据集时的性能。
g SpaDo 在 osmFISH 数据集上的性能,随着丢弃率的增加而增加。红线代表每种方法的原始性能。
h SpaDo 在 DLPFC_151673 数据集上的性能随着丢失率的增加而增加。图的右侧显示了额外丢失的百分比。红线代表每种方法的原始性能
首先,我们评估了 SpaDo 中两个关键参数的鲁棒性:单细胞分辨率空间转录组数据的最近邻数(图 3a)和斑点分辨率空间转录组数据的半径大小(图 3b)。我们发现 SpaDo 在不同的近邻数量(图 3a)和不同的半径大小(图 3b)下都能保持稳健性。值得注意的是,当半径 = 1 时,SpaDo 可能无法充分利用空间信息,除非空间转录组测序方法的光斑半径非常大,如旧的 ST,因此它明显低于其他半径值,这里仅将其作为基线。
其次,SpaDo 依赖于单细胞和点分辨率空间转录组学数据的准确细胞类型注释。在本研究中,SpaDo 对单细胞空间转录组学数据采用 Seurat v4,对点分辨率数据采用 Cell2location [26],以获得这些注释。之所以选择 Cell2location,是因为它在第三方基准测试论文[35]中表现出色。此外,我们还在三个单细胞空间转录组学数据集上验证了 SpaDo 对 Seurat v4 参数进行细胞类型注释的鲁棒性(图 3c)。我们还研究了其他广泛使用的斑点去卷积方法的应用,包括 SPOTlight [36] 和 RCTD [37],用于 12 个斑点分辨率的 DLPFC 切片(图 3d)。我们可以看到,SpaDo 对斑点去卷积方法具有很强的鲁棒性,其中 Cell2location 的中值最高。
第三,我们利用詹森-香农发散(Jensen-Shannon divergence,JSD)来计算 SpaDo 对每个点/单元的 SPACE 距离。为了评估 SpaDo 对不同距离指标的鲁棒性,我们使用了常用指标,如 Spearman 相关性、皮尔逊系数、余弦距离、欧氏距离、曼哈顿距离和 JSD,对单细胞(图 3e,见 "方法")和点分辨率空间转录组学数据(图 3f,见 "方法")进行了性能评估。在这些不同的距离指标中,SpaDo 始终表现出稳健性,其中 JSD 的中值最高。
最后,考虑到空间转录组学数据固有的噪声,我们评估了 SpaDo 和其他现有方法对测序深度和丢失的敏感性。以 osmFISH(单细胞分辨率)和 DLPFC_151673(点分辨率)数据为例,我们通过随机将 10%、30% 和 50% 的非零表达值设为零,人为地增加了丢失率。我们观察到 SpaDo 对较高的丢失率表现出耐受性,而其他方法则明显受到影响(图 3g、h,见 "方法")。
SpaDo 能有效缓解多切片整合中的批次效应
批次效应:是实验或研究中经常遇到的一种现象,它指的是在数据采集过程中由于各种因素导致的系统性变异,而且它与我们研究的变量没有关系!
现有的大多数空间转录组分析方法仅限于单切片空间域分析,因为它们无法整合多个切片的基因表达和空间信息。然而,随着空间转录组学的发展,从同一组织中获取多个切片并对其进行整合以获得新的生物学见解已成为可能 [20,21]。
值得注意的是,最重要的挑战之一在于整合过程中的批次效应。如前所述,SpaDo 利用 SPACE 整合基因表达和空间信息。SPACE 的设计旨在利用细胞类型之间的空间关系,通过各种自动细胞类型注释策略[24, 25, 30]和斑点去卷积方法[14, 15]确保切片之间的一致性。因此,SpaDo 在理论上可以承受批次效应。
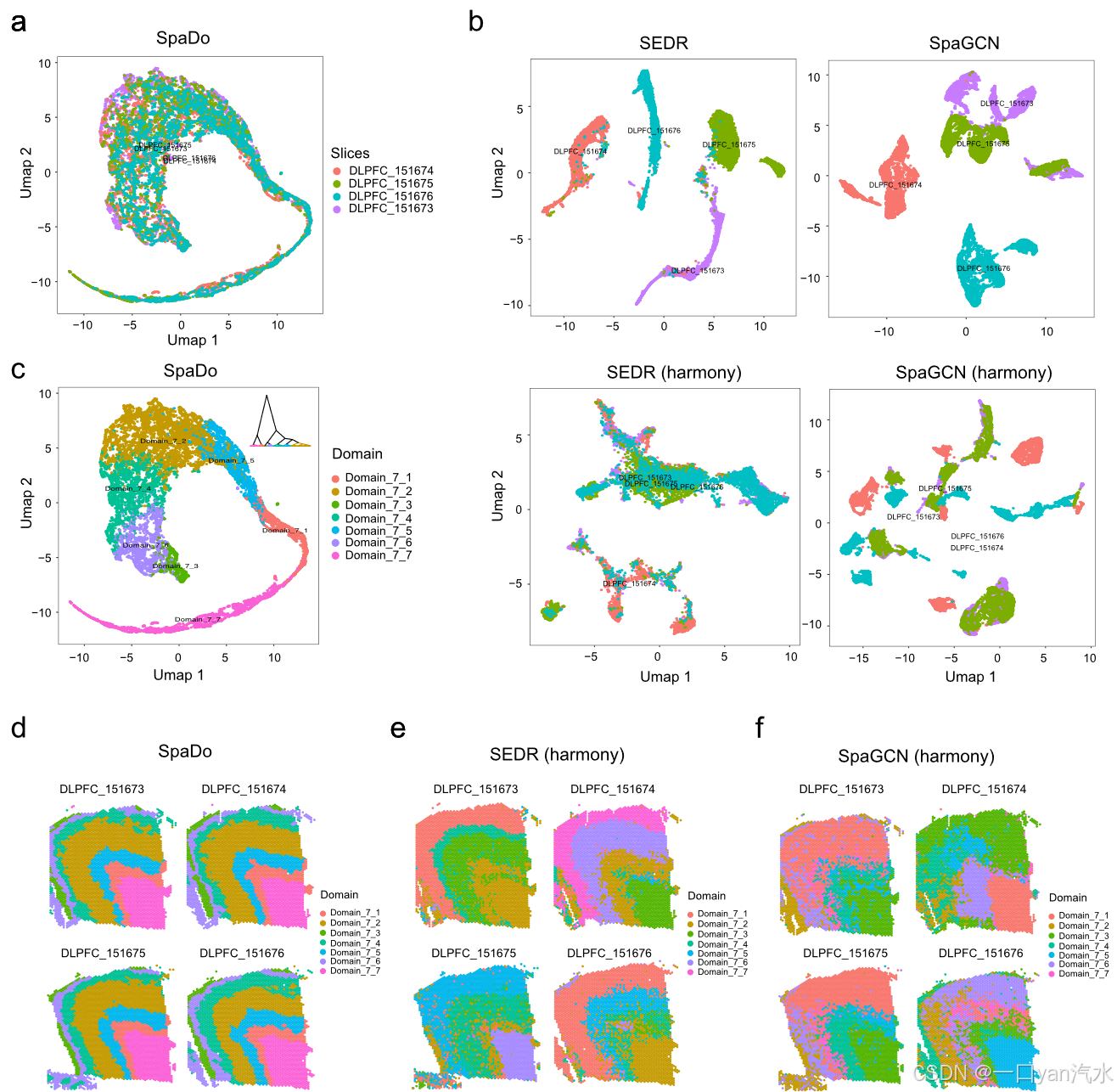
a SpaDo 在四个 DLPFC 切片上的 Umap(以切片为单位着色)。
b 四个 DLPFC 切片上有和谐与无和谐的 SEDR 和 SpaGCN 的 Umap(按切片着色)。
c SpaDo 在四个 DLPFC 切片上的 Umap 和分层聚类结果(按检测到的空间域着色)。
d SpaDo 在四张 DLPFC 切片上检测到的空间域的位置。
e SEDR 在四张 DLPFC 切片上检测到的空间域的位置,其中包含和声。
f SpaGCN 在四张 DLPFC 切片上检测到的空间域的位置,其中包含和声。
为了进一步证明这一点,我们对 SpaDo 和其他现有方法进行了综合比较,以解决单细胞和点分辨率空间转录组学数据的批次效应(图 4 和附加文件 1:图 S9)。对于点分辨率空间转录组学,我们测试了四个 DLPFC 切片,它们属于具有相同七层的同一样本。我们比较了 SpaDo 中使用的 SPACE(图 4a)、SEDR 获得的嵌入和 SpaGCN(有和谐[38]校正批次效应)(图 4c)。结果表明,SpaDo 的空间嵌入(SPACE)能有效减轻多个切片的批次效应。相比之下,SEDR 和 SpaGCN 的嵌入即使与和谐[38]相结合,也不能很好地在多个切片之间保持一致。此外,我们还使用 SpaDo 进行了多切片域检测(图 4c、d)。然后,对于通过 SEDR 和 SpaGCN 组合和谐得到的嵌入,我们采用类似的策略进行多切片域检测(图 4e、f,见 "方法")。我们可以看到,只有 SpaDo 在 4 个 DLPFC 切片之间获得了一致的域结果(图 4d-f)。我们在 3 个 MERFISH 数据上也得到了类似的结论(附加文件 1:图 S9)。
我们通过强调通过其他方法(如 SEDR、SpaGCN 和 STAGATE)生成的内嵌数据仅限于单个数据片内的不同训练空间来澄清这一观点。因此,由这些方法生成的来自不同切片的内嵌值并不一致,即使应用批量校正技术(如和谐)也是如此。相比之下,SPACE 本身以细胞类型的空间分布为基础,这一特征在不同切片中保持一致。SPACE 在表示空间域时具有良好的可解释性,再加上这种一致性,构成了其卓越性能的基础。
SpaDo 可实现多切片空间域检测
我们展示了 SpaDo 在检测空间域方面的实用性,这些空间域可在多个切片之间进行比较,这对于研究跨切片共享空间功能至关重要。
具体来说,我们在多个切片上进行了类似于 TLS 的结构域检测(图 5)。
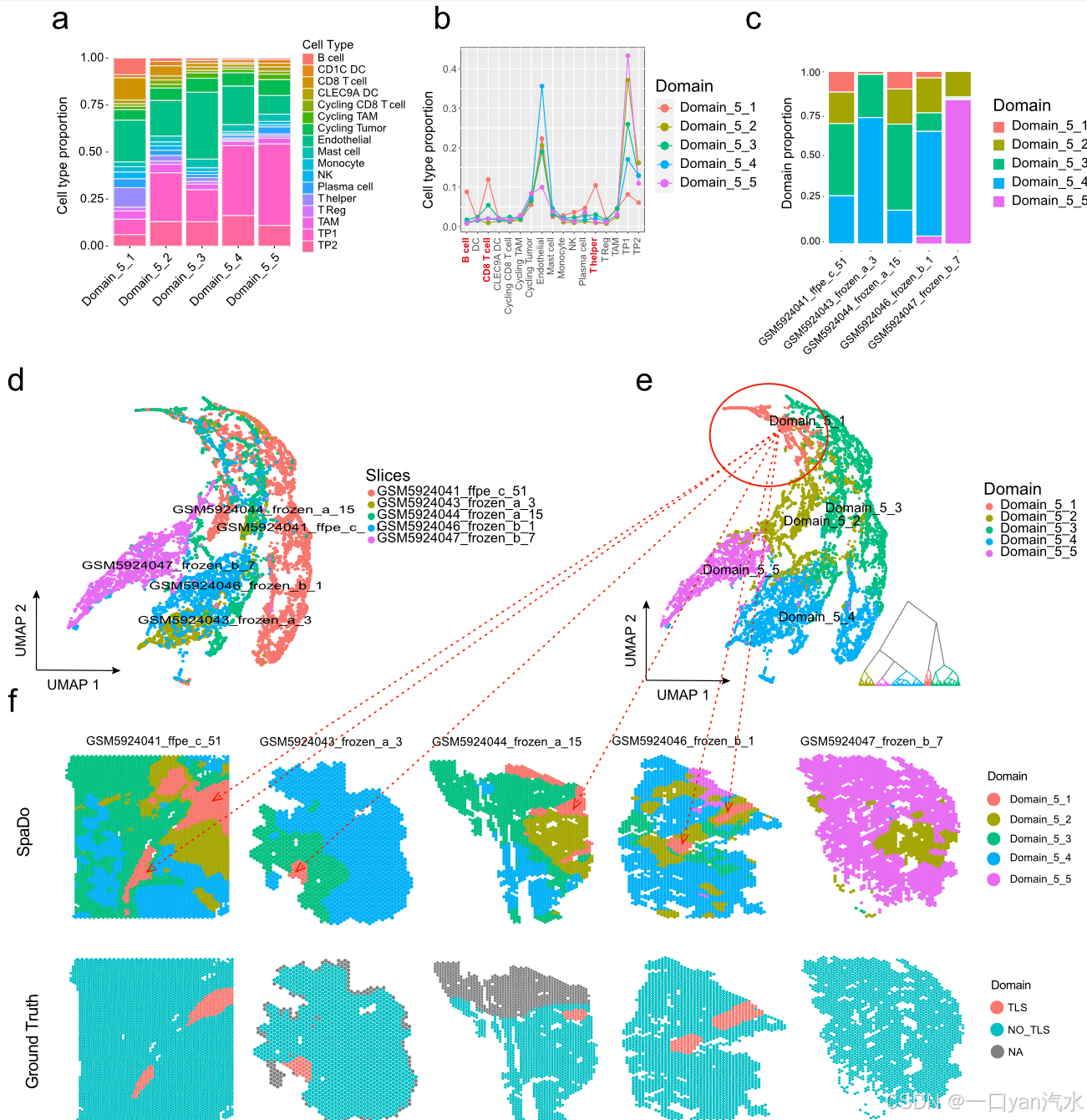
a 每个检测到的域的细胞类型比例。
b 每个检测到的空间域中细胞类型比例的比较。
c 每个 RCC 切片检测到的空间域比例。
d SPACE 对五张 RCC 切片的 Umap 和分层聚类结果(按切片着色)。
e 五张 RCC 切片上 SPACE 的 Umap 和分层聚类结果(按检测到的空间域着色)。
f 所有五个 RCC 切片中检测到的空间域与手动注释的 TLS 标签相对应的位置
我们使用了五张带有人工标注的 TLS 区域的 RCC 切片,其中四张包含一个或两个 TLS 区域,其余一张没有 TLS 区域作为阴性对照[34]。在这项测试中,SpaDo 使用默认的空间域编号选择方法成功检测到了五个空间域(图 5e,见 "方法")。此外,SpaDo 还提供了每个检测到的域中每种细胞类型的确切比例,显示了其良好的可解释性(图 5a)。值得注意的是,在五个空间域中,"Domain_5_1 "显示出 B 细胞、CD8 T 细胞和 T 辅助细胞的显著富集,使其有别于其他四个空间域(图 5b)。此外,"Domain_5_1 "作为共识域在所有四个 TLS 阳性切片中都被检测到,但在阴性对照切片中却没有显示(图 5c)。这些发现表明,"Domain_5_1 "代表了与重要免疫功能相关的常见 TLS 样空间域。Domain_5_1 "在四个切片中的位置与人工标注的 TLS 标签一致(图 5f),进一步证实了 SpaDo 在多个切片中检测共识空间域的能力。这些共识空间域可能是潜在的空间标记。最后,即使将空间域数量设置为 3,也能得到类似的结果,这表明 SpaDo 在检测类似 TLS 的空间域方面具有很强的鲁棒性和很高的灵敏度(附加文件 1:图 S10)。
SpaDo 可实现基于参考的空间域注释 空间域的注释是一项重要任务,但主要由人工完成,费时费力。随着人工注释空间域的空间转录组数据集越来越普遍,SpaDo 可用于自动注释空间域。具体来说,SpaDo 利用这些数据集(被称为带有空间域标签的空间参考)来注释新测序的空间转录组。这一策略在概念上类似于用于序列比对的 BLAST [39]等流行工具,或我们之前使用单细胞转录组参考文献进行自动细胞类型注释的 scLearn 等人的策略 [24、25、30]。就 SpaDo 而言,它通过测量查询细胞的 SPACE 与空间参照中每个空间域中心点之间的距离来执行搜索。然后,根据最小距离将参照中的空间域标签分配给查询细胞(见 "方法")。
这一策略在概念上类似于用于序列比对的 BLAST [39]等流行工具,或我们之前使用单细胞转录组参考文献进行自动细胞类型注释的 scLearn 等人的策略 [24、25、30]。就 SpaDo 而言,它通过测量查询细胞的 SPACE 与空间参照中每个空间域中心点之间的距离来执行搜索。然后,根据最小距离(见 "方法")将参照中的空间域标签分配给查询细胞。然而,PASTE 仅限于相邻切片,因此不适用于多种切片。为了证明 SpaDo 在这项任务中的优越性,我们测试了八个常用的 DLPFC 数据集[33]的空间域注释。每个数据集分别作为参考数据或查询数据处理,结果共有 56 个数据集对(排列 A28 = 56)(图 6a,见 "方法")。结果清楚地表明,与 Seurat v4 和 PASTE 相比,SpaDo 获得了更高的宏 F1 分数(图 6a)。此外,我们还使用 "DLPFC_151673 "作为空间参考,并使用其余七个 DLPFC 数据集作为查询,对这些结果进行了说明。SpaDo 为 "DLPFC_151673 "计算的 UMAP 空间可视化与人工标注的空间域标注高度一致(图 6b,c)。此外,在所有查询数据集上,SpaDo 的注释准确性都优于 Seurat v4(图 6d、f)。另一方面,PASTE 在 7 个数据集中的 5 个数据集上几乎都失败了,它将所有斑点都预测为属于同一空间域(图 6g)。
SpaDo 支持多切片聚类分析
多切片聚类分析在研究发育状况的空间异质性方面起着至关重要的作用[40, 41]。进行多切片聚类分析的关键在于计算不同切片的相似性。传统上,测量多个切片的全局相似性是基于相似切片具有相似细胞类型组成的假设,而不考虑空间信息。然而,SpaDo 采用了不同的视角,假设如果两个切片相似,它们的空间域组成(图 1b 和 7f,见 "方法")也相似,反之亦然。这使得 SpaDo 能够正确地进行多切片聚类分析。
因此,我们利用三项空间转录组学研究[4042]进行了比较,以评估 SpaDo 在多切片聚类分析方面的性能。第一项研究的重点是构建发育中人类心脏的时空细胞图谱,利用发育中人类心脏在三个发育阶段(即受孕后 5、6 和 9 周(PCW))的 19 个空间转录组(ST)切片(图 7)。我们首先使用 SpaDo 进行了多切片空间域检测(图 7a)。具有相同时间点的数据集往往聚集在一起,这表明 SpaDo 能有效捕捉每个切片的底线信息,而不受批次效应的影响(图 7b)。据观察,同一时间点的切片表现出相似的空间域构成(图 7d)。此外,与基线方法(根据细胞类型组成计算切片相似度)相比,SpaDo 显示出更一致的聚类结果(图 7e、f,见 "方法")。重要的是,这种改进的性能在不同的选定域数中都是稳健的(附加文件 1:图 S11)。另外两个数据集,即发育中的鸡心脏数据集[42](附加文件 1:图 S12 和 13)和人类皮质类器官数据集[41](附加文件 1:图 S14)也得到了类似的结果。这些发现凸显了 SpaDo 在多切片聚类中有效整合空间信息以测量多切片全局相似性的能力。
讨论
随着空间转录组的发展,多个组织切片的积累越来越多,并可进一步整合以揭示转录组和细胞景观的新见解。然而,挑战在于如何有效整合基因表达和空间信息,使其在多个切片中既可解释又可比较。目前的策略仅限于单切片领域分析,计算复杂度相对较高,可解释性有限。因此,我们提出 SpaDo 作为一个高效的框架,旨在促进单细胞和斑点分辨率的多切片空间域分析。
SpaDo 通过三个关键应用进行空间转录组学分析:多切片空间域检测、基于参考的空间域注释和多切片聚类分析。通过对来自不同生物背景和测序平台的 40 多个不同空间切片的检测,我们证明了 SpaDo 对不同参数的鲁棒性和对噪声的耐受性(图 3)。此外,SpaDo 还能有效解决批次效应,无需额外校正。总之,SpaDo 极大地增强了空间转录组学数据的分析能力,尤其是在涉及多切片空间域的情况下。
SpaDo 的关键创新在于设计了一种简单而有效的嵌入方法,即 SPACE。通过将细胞类型注释与空间龛位相结合,SPACE 成功地整合了多个切片的基因表达和空间信息,表现出了对高噪声和批次效应的耐受性(图 3 和图 4)。我们强调了 SPACE 优于其他复杂空间嵌入方法(如 SEDR、SpaGCN 和 STAGATE)的三个主要原因:(1)SPACE 与空间域的生物学特征非常吻合,因而具有良好的可解释性。(2) SPACE 有效地解决了批处理效应。(3) 不同空间域之间的差异相对明显,不需要特别精细的空间信息来区分它们[31]。因此,尽管 SPACE 可能会平滑特征并减少组织异质性,但它在多切片空间域分析中仍然表现出色。在我们的研究中,我们没有观察到 SPACE 简化了空间结构(图 S4 和 S6,以及图 2e)。我们推测这可能有三个原因。首先,虽然单个细胞水平的空间信息可能会丢失,但基本的空间信息--特别是整个空间域的细胞类型组成--得到了有效保留。其次,平滑操作固有的抗噪性有助于通过 SPACE 嵌入消除空间转录组数据中的噪声。最后,最近的研究[31, 32]证明,空间域之间的明显区别并不需要复杂的细节。总之,在空间域检测中,SPACE 在噪声容限和特征平滑之间保持了微妙的平衡,因此非常适合整合多个切片。
SpaDo 有三个潜在的局限性。首先,它依赖于 Seurat v4 和 Cell2location 等细胞类型标注方法。不过,即使在细胞类型标注方法表现略微不理想的情况下,SpaDo 仍能显示出相对有利的结果(图 3c、d)。其次,虽然我们已经证明由 SpaDo 设计的 SPACE 非常适合空间域分析,但它可能倾向于平滑特征并降低组织的异质性。因此,在应用 SPACE 进行空间域以外的分析时应谨慎。第三,SpaDo 专注于多切片的空间域分析,可能不适合细胞级三维组织重建。
虽然我们的研究主要侧重于将 SpaDo 应用于空间转录组数据,但值得注意的是,SpaDo 有可能扩展到多模态空间数据分析。如果能识别不同 omics 数据集中的相应细胞类型,这种扩展就会特别有价值。值得注意的是,空间表观基因组学[43, 44]和滑动 DNA-seq 等技术[45]的进步为将表观遗传学和 DNA 信息整合到空间分析中提供了令人兴奋的机会。随着空间 DNA-seq 技术(如 slide-DNA-seq)的不断发展,它们有可能破解更准确的肿瘤演变模式[46]。而 SpaDo 正准备利用这些发展,将来自多个切片的空间多模态数据整合起来。这种整合有望发现新的肿瘤标记物,包括一致的和不同的肿瘤演变模式,从而有助于加深对空间生物学的理解。
结论
总之,SpaDo 是空间转录组学多切片空间域分析的开创性框架。它在检测多切片空间域、提供基于参考的空间域注释以及进行多切片聚类分析方面表现出色,解决了单切片域分析的局限性。SPACE 嵌入确保了良好的可解释性、较强的鲁棒性和较高的噪声容忍度,使 SpaDo 成为研究人员的重要空间转录组学分析工具。

















 764
764

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









