




 该文详细介绍了对预处理后的单细胞RNA测序数据进行分析的步骤,包括使用Seurat包进行数据整合、细胞注释、细胞类型手动注释、细胞浸润差异分析以及Hallmarker功能差异分析。通过对不同组织的细胞类型比例进行统计,揭示了细胞类型的分布差异,并通过功能富集分析挖掘了特定细胞类型的信号通路差异。
该文详细介绍了对预处理后的单细胞RNA测序数据进行分析的步骤,包括使用Seurat包进行数据整合、细胞注释、细胞类型手动注释、细胞浸润差异分析以及Hallmarker功能差异分析。通过对不同组织的细胞类型比例进行统计,揭示了细胞类型的分布差异,并通过功能富集分析挖掘了特定细胞类型的信号通路差异。
针对(一)已经完成预处理后的数据,这里我采用了两套数据进行分析,由于涉及毕业设计内容,采用G1和G2代替预处理后的两套数据。下面展开对数据的注释、简单的浸润差异分析与Hallmarker功能差异分析。
第一步:数据读入
对(一)保存的数据进行读入,这里共两套数据
G1 <- readRDS(file = './G1_obj.RDS') G2 <- readRDS(file = './G2_obj.RDS')
第二步:添加额外信息(meta信息)
需要对单细胞的meta信息进行添加的才进行这一步骤。这里对G1进行添加,如G2需要,步骤同下
Meta<- read.csv("./G1_Meta.csv") Meta$Dataset <- "G1" colnames(Meta)[1] <- "orig.ident" row_name <- rownames(G1@meta.data) G1@meta.data$id <- 1:nrow(G1@meta.data) G1@meta.data <- merge(G1@meta.data, Meta, by = "orig.ident") G1@meta.data <- G1@meta.data[order(G1@meta.data$id ),] rownames(G1@meta.data) <- row_name head(Meta,1)注:这里使用merge会打乱数据框原有的顺序,因此采用提前添加id,之后进行排序完成顺序的校正,从而补上merge后缺失的行名
第三步:合并数据,并进行细胞自动注释与手动注释
合并数据并自动注释
#合并数据,并进行预处理 BRCA_SingleCell.T <- merge(G1, G2) rm(G1) rm(G2) BRCA_SingleCell.T <- NormalizeData(BRCA_SingleCell.T, normalization.method = "LogNormalize") BRCA_SingleCell.T <- FindVariableFeatures(BRCA_SingleCell.T, selection.method = "vst", nfeatures = 3000) BRCA_SingleCell.T <- ScaleData(BRCA_SingleCell.T, vars.to.regress = c('nCount_RNA')) BRCA_SingleCell.T <- RunPCA(BRCA_SingleCell.T) BRCA_SingleCell.T <- RunHarmony(object = BRCA_SingleCell.T,group.by.vars = c('Dataset')) BRCA_SingleCell.T <- RunUMAP(BRCA_SingleCell.T,reduction = "harmony",dims = 1:30,seed.use = 12345) BRCA_SingleCell.T <- FindNeighbors(BRCA_SingleCell.T,reduction = 'harmony', dims = 1:30, verbose = FALSE) BRCA_SingleCell.T <- FindClusters(BRCA_SingleCell.T,resolution = 0.5, verbose = FALSE,random.seed=20220727) #对合并数据重新进行细胞自动注释 cellAnnotation <- function(obj,markerList,assay='SCT',slot='data'){ markergene <- unique(do.call(c,markerList)) Idents(obj) <- 'seurat_clusters' cluster.averages <- AverageExpression(obj,assays=assay, slot = slot,features=markergene, return.seurat = TRUE) if(assay=='SCT'){ scale.data <- cluster.averages@assays$SCT@data }else{ scale.data <- cluster.averages@assays$RNA@data } print(scale.data) cell_score <- sapply(names(markergeneList),function(x){ tmp <- scale.data[rownames(scale.data)%in%markergeneList[[x]],] if(is.matrix(tmp)){ if(nrow(tmp)>=2){ res <- apply(tmp,2,max) return(res) }else{ return(rep(-2,ncol(tmp))) } }else{ return(tmp) } }) print(cell_score) celltypeMap <- apply(cell_score,1,function(x){ colnames(cell_score)[which(x==max(x))] },simplify = T) obj@meta.data$cellType_auto <- plyr::mapvalues(x = obj@active.ident, from = names(celltypeMap), to = celltypeMap) return(obj) } lymphocyte <- c('CD3D','CD3E','CD79A','MS4A1','MZB1') myeloid <- c('CD68','CD14','TPSAB1' , 'TPSB2','CD1E','CD1C','LAMP3', 'IDO1') EOC <- c('EPCAM','KRT19','CD24') fibo_gene <- c('DCN','FAP','COL1A2') endo_gene <- c('PECAM1','VWF') markergeneList <- list(lymphocyte=lymphocyte,myeloid=myeloid,Epi=EOC,fibo=fibo_gene,endo=endo_gene) BRCA_SingleCell.T <- cellAnnotation(obj=BRCA_SingleCell.T,assay = 'RNA',markerList=markergeneList)合并之后可视化合并后的数据
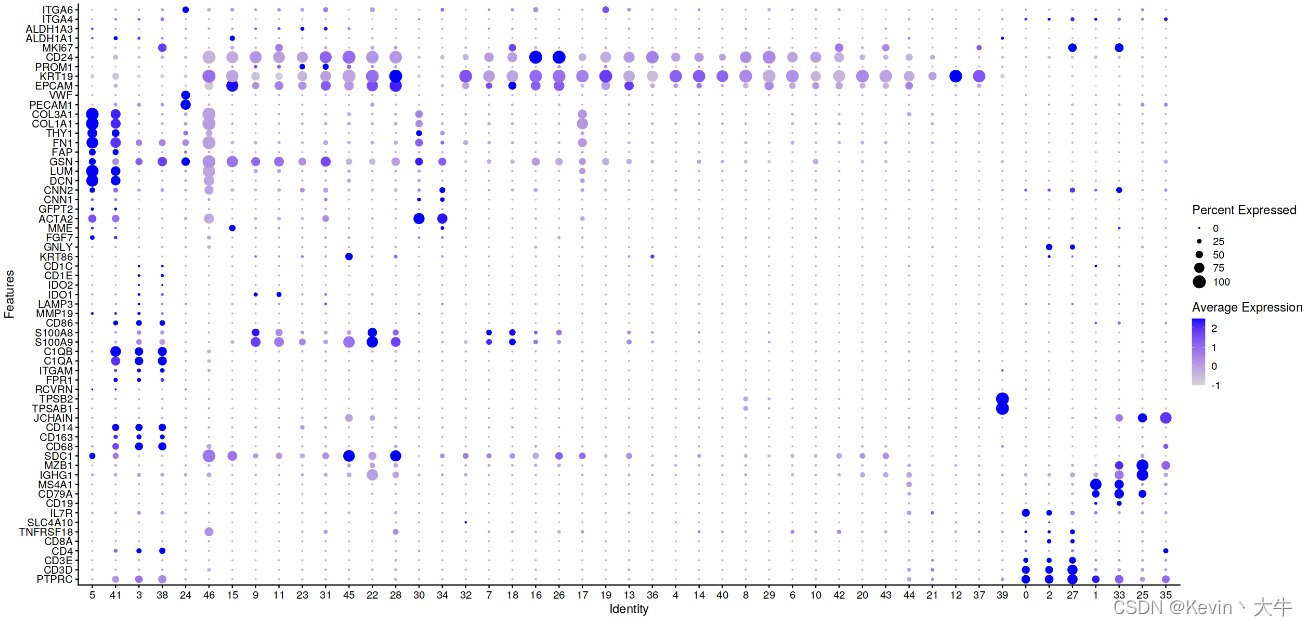
展示细胞簇的基因表达情况
genes_to_check = c('PTPRC', 'CD3D', 'CD3E', 'CD4','CD8A','TNFRSF18','SLC4A10','IL7R', 'CD19', 'CD79A', 'MS4A1' , 'IGHG1', 'MZB1', 'SDC1', 'CD68', 'CD163', 'CD14', 'JCHAIN', 'TPSAB1' , 'TPSB2', 'RCVRN','FPR1' , 'ITGAM' , 'C1QA', 'C1QB', 'S100A9', 'S100A8','CD86', 'MMP19', 'LAMP3', 'IDO1','IDO2', 'CD1E','CD1C', 'KRT86','GNLY', 'FGF7','MME','ACTA2','GFPT2','CNN1','CNN2', 'DCN', 'LUM', 'GSN' , 'FAP','FN1','THY1','COL1A1','COL3A1', 'PECAM1', 'VWF', 'EPCAM' , 'KRT19', 'PROM1', 'CD24','MKI67', 'ALDH1A1', 'ALDH1A3','ITGA4','ITGA6') DotPlot(BRCA_SingleCell.T,group.by = 'seurat_clusters', features = unique(genes_to_check),cluster.idents = T) + coord_flip()结果如下:
根据基因表达图,手动注释细胞类型
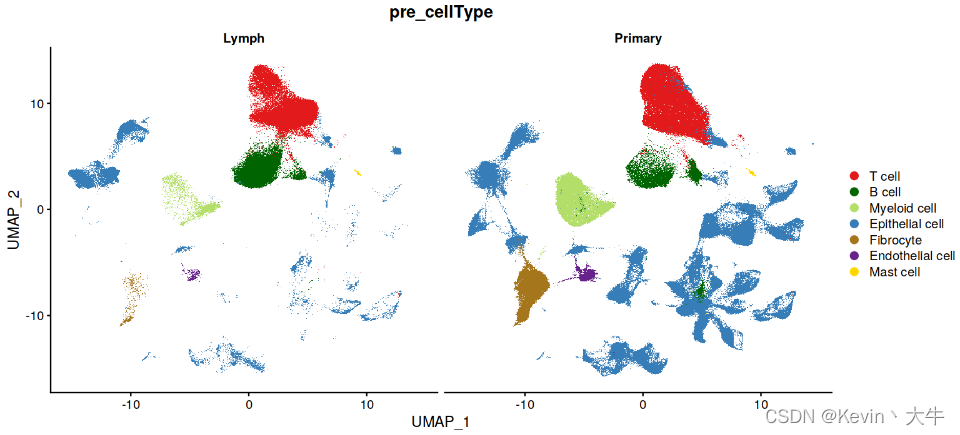
B_P_cell=c(1,25,36) Mast_cell=c(39) T_cell=c(0,2) Epithelial=c(14,10,11,23,31,46,21,28,30,35,32,7,18,16,26,17,19,43,45,22,12,33,42,20,44,8,29,6,9,4,15,40,13,37) Endothelial =c(24) Myeloid=c(3) Fibro_cell=c(5,47) MIX=c(38,27,34,41) table(c(B_P_cell,Mast_cell,T_cell,Epithelial,Endothelial,Myeloid,Fibro_cell,MIX)) print(setdiff(0:47,c(B_P_cell,Mast_cell,T_cell,Epithelial,Endothelial,Myeloid,Fibro_cell,MIX))) current.cluster.ids <- c(B_P_cell,Mast_cell,T_cell,Epithelial,Endothelial,Myeloid,Fibro_cell,MIX) new.cluster.ids <- c(rep("B cell",length(B_P_cell)), rep("Mast cell",length(Mast_cell)), rep("T cell",length(T_cell)), rep("Epithelial cell",length(Epithelial)), rep("Endothelial cell",length(Endothelial)), rep("Myeloid cell",length(Myeloid)), rep("Fibrocyte",length(Fibro_cell)), rep("MIX",length(MIX)) ) BRCA_SingleCell.T@meta.data$pre_cellType <- plyr::mapvalues(x = BRCA_SingleCell.T$seurat_clusters, from = current.cluster.ids, to = new.cluster.ids) BRCA_SingleCell.T <- subset(BRCA_SingleCell.T, pre_cellType %in% c("T cell","B cell","Myeloid cell","Epithelial cell","Fibrocyte", "Endothelial cell","Mast cell"))
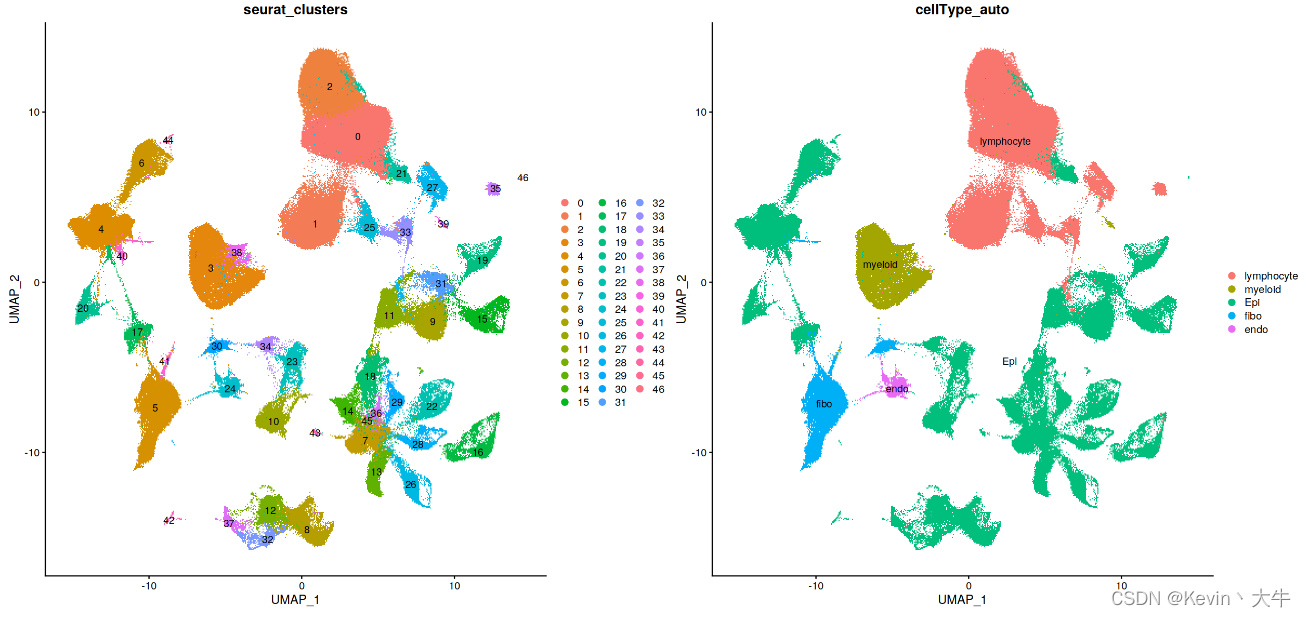
第四步:细胞浸润差异分析
展示不同组织细胞类型的UMAP图,可以发现两个组织之间的细胞浸润是存在差异的,于是我们针对这个现象进行统计分析
(1)首先进行CellMarker的展示,证明细胞类型之间具有明显的区分度
Idents(BRCA_SingleCell.T) <- 'pre_cellType' cellType_markerGene <- FindAllMarkers(BRCA_SingleCell.T,logfc.threshold = 0.5,only.pos = T,test.use = 'roc',min.pct = 0.2) cellType_markerGene <- subset(cellType_markerGene,!grepl(pattern = 'RP[LS]',gene)) cellType_markerGene <- subset(cellType_markerGene,!grepl(pattern = 'MT-',gene)) top5 <- cellType_markerGene %>% group_by(cluster) %>% top_n(n = 10, wt = myAUC) print(top5) B_P_gene <- c('MS4A1','CD79A','IGHG1','JCHAIN') T_gene <- c('CD3D','CD3E') Epithelial_gene <- c('EPCAM','CD24') Endothelial_gene <- c('PECAM1','VWF') Myeloid_gene <- c('CD68','CD14') Mast_gene <- c('TPSAB1','TPSB2') Fibro_gene <- c('DCN','LUM') cellMarker <- c(B_P_gene,T_gene,Epithelial_gene,Endothelial_gene,Myeloid_gene, Mast_gene,Fibro_gene) BRCA_SingleCell.T$pre_cellType <- factor(BRCA_SingleCell.T$pre_cellType,levels=c("B cell","T cell","Epithelial cell","Endothelial cell","Myeloid cell","Mast cell","Fibrocyte")) DotPlot(BRCA_SingleCell.T,group.by = 'pre_cellType',cols = c('blue','red'), features = unique(cellMarker),cluster.idents = F) + t
 最低0.47元/天 解锁文章
最低0.47元/天 解锁文章



















 2503
2503

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











