



翻译重编程是黑色素瘤中表型可塑性和治疗抗性的进化保守驱动因素
保拉·法莱塔,1,10 路易斯·桑切斯‐德尔坎波,1,10 贾加特·乔汉,1 迈克·埃弗恩,2 艾米·肯永,3 克里斯托弗·J·科尔肖,4 罗伯特·西达韦,1 理查德·莱斯尔,1 拉思穆斯·弗雷特,1 马修·J·丹尼尔斯,5 卢欣,1 托马斯·图廷,6 马克·米德尔顿,7 弗朗西斯卡·M·布法,7 安妮·E·威利斯,8 格雷厄姆·帕维特,4 泽耶夫·A·罗奈,9 塔季扬娜·绍卡‐施彭勒,3 迈克尔·赫尔策尔,2 和科林·R·戈丁1
1 路德维希癌症研究所,纳菲尔德临床医学系,牛津大学,亨丁顿,牛津 OX3 7DQ,英国; 2 临床化学与临床药理学系,RNA生物学单元,波恩大学医院,德国波恩 D‐53127,德国; 3 韦瑟罗尔分子医学研究所,拉德克利夫医学系,牛津大学,约翰·拉德克利夫医院,亨丁顿,牛津 OX3 9DS,英国; 4 分子与细胞功能部,生物科学学院,生物、医学与健康学院,曼彻斯特大学,曼彻斯特 M13 9PT,英国; 5 心血管医学部,拉德克利夫医学系,牛津大学,约翰·拉德克利夫医院,亨丁顿,牛津 OX3 9DU,英国; 6 实验皮肤病学实验室,皮肤科与过敏科,马格德堡大学医院,德国马格德堡 39120,德国; 7 肿瘤科,牛津大学,亨丁顿,牛津 OX3 7DQ,英国; 8 医学研究理事会毒理学部门,莱斯特 LE1 9HN,英国; 9 肿瘤起始与维持项目,癌症中心,桑福德‐伯纳姆‐普雷比斯医学发现研究所,拉霍亚,加利福尼亚州 92037,美国
肿瘤内微环境产生表型不同但可相互转化的恶性细胞亚群,从而促进转移扩散和治疗抗性。不同的微环境信号是否通过共同机制诱导侵袭性或治疗抵抗表型尚不清楚。在黑色素瘤中,谱系存活癌基因小眼症相关转录因子(MITF)的低表达与侵袭、衰老和耐药性相关。然而,MITF在体内如何被抑制,以及肿瘤中的MITF低表达细胞如何逃逸衰老,仍知之甚少。本文表明,包括介导对过继性T细胞免疫疗法抵抗的炎症在内的微环境信号,可通过ATF4在翻译起始因子 eIF2B受到抑制时转录抑制MITF。ATF4作为整合应激反应的关键转录中介,还能激活AXL并抑制衰老,从而形成在人类肿瘤中观察到的MITF低/AXL高药物抗性表型。然而,出乎意料的是,在没有翻译重编程的情况下,ATF4高/ MITF低状态不足以驱动侵袭。重要的是,翻译重编程显著增强肿瘤发生,并与先前无法解释的抗PD‐1免疫治疗抵抗相关基因表达程序相关联。由于我们发现eIF2B的抑制也能驱动神经嵴迁移和酵母侵袭性,因此结果提示,翻译重编程作为一种进化保守的饥饿反应,已被黑色素瘤中的微环境应激信号劫持,以驱动表型可塑性和侵袭,并决定治疗结局。
[关键词:黑色素瘤;表型转换;侵袭性;MITF;TNFα]
本文的补充材料可获取。
2016年9月19日收到;修订版于2016年12月21日接受
多年来,癌症进展(包括耐药性产生)的遗传基础已变得越来越明确。然而,在肿瘤的异质性遗传景观之上还存在着其他因素
肿瘤内微环境的影响。多种微环境信号的变化,包括缺氧、营养水平、来自浸润性免疫细胞的信号以及间质信号共同作用,诱导特定的基因表达程序,从而决定侵袭潜力或治疗反应。然而,耐药和免疫治疗耐药表型如何与侵袭性相关,以及多种肿瘤内微环境线索是否汇聚于一个 “通用”的表型可塑性调控因子,目前仍知之甚少。
黑色素瘤是一种以早期转移能力著称的癌症,是解析侵袭、耐药性和免疫治疗耐药性之间复杂关系的优秀模型。对86种黑色素瘤细胞系进行基因表达谱分析(Hoek 等, 2006)发现,其基因表达特征反映了两种表型之一:具有侵袭性但增殖能力差,或快速增殖但侵袭性差。侵袭性和增殖性特征之间的负相关尚不完全清楚,但与黑色素细胞谱系的关键调控因子——小眼症相关转录因子(MITF)的表达相关(Hodgkinson 等,1993)。MITF是一种谱系存活癌基因(Garraway 等,2005),可驱动增殖( Carreira 等,2006)、促进存活,并激活参与黑色素分化相关生成的基因。相反,MITF低表达细胞为慢周期且具有侵袭性(Carreira 等,2006;Cheli 等,2010)。然而,短期MITF耗竭会导致侵袭性增加并获得肿瘤起始表型( Cheli 等人 2011b),而长期MITF耗竭则导致衰老( Giuliano等,2010)。然而,体内衰老主要局限于痣( Michaloglou 等,2005;Gray‐Schopfer 等,2006), 尽管黑色素瘤中可能含有大量MITF阴性细胞(Goodall 等, 2008;Riesenberg等人2015)。在体内,MITF如何被下调而不引发衰老仍不清楚,但低MITF水平与对MEK和 BRAF抑制剂的耐药性相关(Konieczkowski等,2014; Muller等,2014;Dugo等,2015)。因此,解析体内产生MITF低表达非衰老细胞的分子机制,对于理解侵袭、衰老旁路和治疗耐药性之间的复杂关系至关重要。
在此,我们发现MITF受到触发翻译起始因子eIF2B抑制的信号的翻译抑制,并受到整合应激反应(ISR)关键组成部分、一种翻译调控的转录因子ATF4的转录抑制。我们揭示了翻译重编程是黑色素瘤表型可塑性的主调控因子,决定了药物和免疫治疗耐药性以及转移潜能。
结果
谷氨酰胺限制抑制MITF
为了初步了解肿瘤内微环境如何产生MITF低表达细胞,我们重点关注了谷氨酰胺的潜在作用。谷氨酰胺作为一种条件必需氨基酸,可作为合成代谢的碳和氮来源,在实体瘤中通常被耗尽(Roberts 等,1956; Kamphorst 等,2015)。尽管谷氨酰胺可以进行从头合成,但许多细胞,尤其是癌细胞(Wise 和 Thompson,2010),包括黑色素瘤(Wang 等,2014),都需要其外源性供给,而谷氨酰胺是黑色素瘤肿瘤核心中五种关键耗竭氨基酸之一(Pan 等,2016)。此外,BRAF抑制剂耐药的黑色素瘤表现出强烈的谷氨酰胺成瘾(Hernandez‐Davies 等, 2015;Baenke 等,2016)。
将黑色素瘤细胞从’杜氏改良伊格尔培养基(DMEM)转移至缺乏谷氨酰胺、丝氨酸和甘氨酸的最低必需培养基 (MEM)后,人SKmel28、小鼠B16(图1A)或人 IGR37(补充图S1A)黑色素瘤细胞系中的MITF表达均降低。添加谷氨酰胺可恢复MITF表达,而添加丝氨酸或甘氨酸则不能。需要注意的是,在通过蛋白质印迹法分析的大多数黑色素瘤细胞系中,MITF表现为两条条带,其中上层条带对应于ERK在Ser73位点对MITF的磷酸化( Hemesath 等,1998),此前已证明该修饰可增强 MITF’的转录活性( Price 等,1998)。谷氨酰胺限制导致的MITF减少具有特异性,因为PAX3和BRN2这两种黑色素细胞谱系转录因子并未受到抑制(图1B)。转移至MEM还引发了MITF磷酸化(图1B),该过程由ERK介导,因为它与磷酸化ERK水平升高相关(补充图S1B,左图),并且可通过使用 MEK抑制剂U0126加以封闭(补充图S1B,右图)。重要的是,谷氨酰胺滴定实验(图1C)显示,在2 mM谷氨酰胺条件下MITF表达得以维持,相较于DMEM中的4 mM谷氨酰胺,但在IGR37细胞中,当谷氨酰胺浓度降至0.5 mM (血液中谷氨酰胺的生理浓度)时,MITF表达即下降。在所有细胞系中,当谷氨酰胺浓度为0.1 mM时,MITF表达受到严重抑制。
谷氨酰胺剥夺期间MITF mRNA出现短暂增加(图1 D),这与CREB(环磷酸腺苷反应元件结合蛋白)磷酸化水平升高相关(图1E),而CREB是MITF表达的已知调控因子(Bertolotto 等,1996)。CREB的修饰可被广谱钙调蛋白拮抗剂氯丙嗪(补充图S1C,上图)或钙依赖性钙调蛋白激酶II抑制剂KN‐93(补充图S1C,下图)所抑制,这与CAMKII激活CREB的结果一致(Ma 等,2014)。氯丙嗪也封闭了谷氨酰胺剥夺引起的MITF mRNA表达的早期上升(补充图S1D),表明CREB磷酸化促进了MITF启动子活性的增强。H89(一种cAMP激活的蛋白激酶A抑制剂)则不能封闭CREB磷酸化(数据未显示)。综上所述,在早期阶段,谷氨酰胺剥夺通过Ca2+依赖的CaMKII介导的 CREB磷酸化短暂增加MITF mRNA表达,而ERK介导的 MITF磷酸化将增强其转录活性(Price 等,1998)。然而,从急性期到长期谷氨酰胺剥夺的转变过程中,MITF呈现动态调控,后期其水平下降。
一个转录翻译偶联的ATF4–MITF反馈环路
使用19,982个探针对16,118个基因进行分析,结果显示,在谷氨酰胺饥饿处理6、24和72小时后,IGR37黑色素瘤细胞的 mRNA中共有4336个差异表达下调基因(5003个探针)并证实MITF下调(补充图S2A; 补充表S1)。对MITF结合基因进行无监督层次聚类(补充图S2B;补充表S2;Strub等人2011年)发现共调控基因簇 (第1–7组)。已知的MITF靶基因,包括许多参与黑色素体功能的基因(图2A),位于第6组且表达下调,表明谷氨酰胺限制促进去分化。然而,我们未观察到谷氨酰胺剥夺对色素沉着的影响,最可能的原因是细胞在G1期聚集(见下文),因此无法通过细胞分裂稀释已有的黑色素体。
在谷氨酰胺饥饿后差异调控的基因中,其中一个最引人关注的是ATF4。ATF4转录因子是综合应激反应(ISR)的关键介质(Harding等人2003年),可调节有助于缓解营养应激的基因,例如通过上调营养物质转运蛋白和参与自噬的基因。MITF磷酸化与编码ATF4的信使RNA增加相关(图2A;补充图S2C)。由于MITF直接调控ATF4启动子荧光素酶报告基因(补充图S2D)并结合ATF4启动子 (补充图S2E),我们的结果表明,在谷氨酰胺饥饿后的早期阶段,MITF激活可能通过上调ATF4促进综合应激反应。
ATF4’的翻译在营养丰富的条件下受到抑制,但在应激触发翻译起始因子eIF磷酸化时会增强2α(Harding等人 2003年)。在谷氨酰胺饥饿条件下,eIF的磷酸化和 ATF4表达增加(补充图S2F),且在ATF4积累之前即可检测到p‐eIF(图2B)。ATF4表达受谷氨酰胺抑制,但不受丝氨酸或甘氨酸抑制(补充图S2G)。硫氧还蛋白( Thapsigargin)作为一种强效的内质网应激诱导剂,被用作阳性对照。在0.2 mM谷氨酰胺条件下,ATF4被诱导而 MITF被抑制(补充图S2H);在此浓度下,谷氨酰胺在2小时内诱导ATF4,并在24小时内抑制MITF(补充图S2I)。
谷氨酰胺限制条件下ATF4的增加与对其靶基因的调控相关(图2C;补充图S2J;补充表S3;Han et al. 2013),同时MITF mRNA水平下降(图2C)。值得注意的是,在三种经改造可于营养丰富条件下表达多西环素诱导型 ATF4的细胞系中,ATF4抑制了MITF的表达(图2D;补充图S2K),同时也抑制了参与黑色素生成的MITF靶基因 TYR和MART1(图2D)。异位ATF4表达还抑制了 MITF启动子荧光素酶报告基因(图2E),表明其可能直接作用于MITF转录,并诱导G1期细胞周期阻滞(图2F),该效应与MITF沉默所介导的阻滞相似(Carreira等, 2006)。然而,尽管siRNA介导的ATF4耗竭在很大程度上消除了谷氨酰胺饥饿条件下对MITF mRNA的抑制(图2 G,左图),但MITF蛋白表达的抑制仍不受影响(图2G,右图),表明MITF的抑制是由第二种ATF4非依赖性机制介导的。这很可能是MITF翻译的抑制,因为MEM培养导致降低的
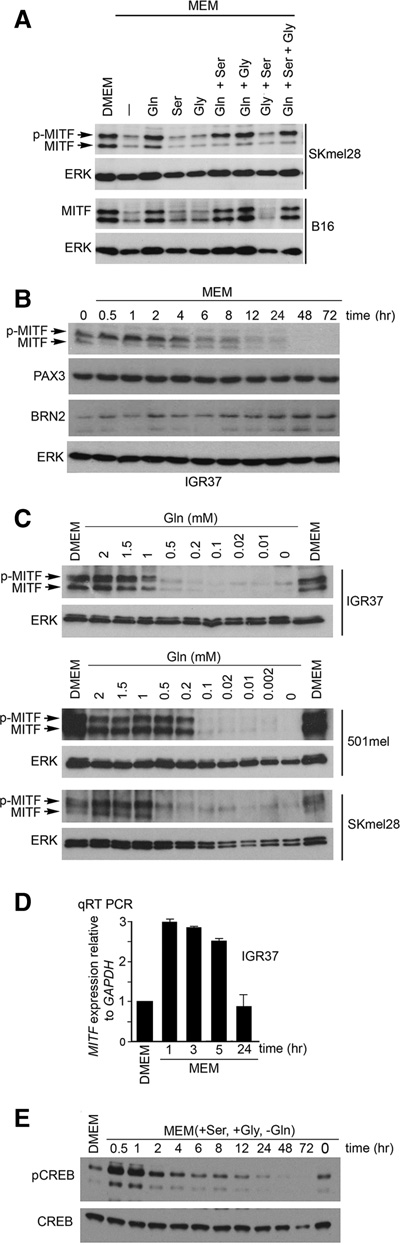
35蛋白质中S‐甲硫氨酸的掺入(最可能是由于eIF2a磷酸化增加),在添加谷氨酰胺后得以恢复,但丝氨酸和甘氨酸无法实现这一效果(补充图S2L,左图)。MITF的免疫沉淀显示,除非存在谷氨酰胺,否则未检测到蛋白质中S‐甲硫氨酸的掺入。考马斯亮蓝染色用作对照(补充图S2L,右图)。
综上所述,这些数据表明,MITF抑制可能通过p‐eIF2α介导的翻译抑制以及ATF4介导的转录抑制共同实现。总体而言,我们的结果表明,翻译重编程及ATF4诱导耦合信号激活综合应激反应,进而通过MITF抑制驱动黑色素瘤去分化。
接下来,我们从103个基因中推导出一个谷氨酰胺饥饿特征(GSS)(补充表S4),这些基因在谷氨酰胺剥夺6小时后表达发生显著变化。选择6小时的时间点是为了避免后期可能出现的谷氨酰胺限制的间接效应。利用GSS基因的平均表达水平对癌症基因组图谱(TCGA; http://cancergenome.nih.gov)中471个人类黑色素瘤样本的基因表达数据进行评分排序。ATF4及其靶基因AS NS的表达随GSS评分呈相应变化(图2H,上图);GSS评分较低的黑色素瘤表现出较低的ATF4/ASNS平均表达水平,而GSS评分较高的黑色素瘤则表现出更高的ATF4/ASNS表达水平。相反,MITF或其分化相关靶基因MLANA的表达与GSS评分呈负相关(图2H,下图)。从机制上讲,谷氨酰胺限制启动了一个转录/翻译偶联的反馈环路:MITF首先增加ATF4 mRNA,eIF2α磷酸化促进ATF4的翻译,而 ATF4则在转录水平上抑制MITF,同时MITF的翻译也被阻断,这很可能是因为p‐eIF2α介导的eIF2B抑制所致(图2I)。
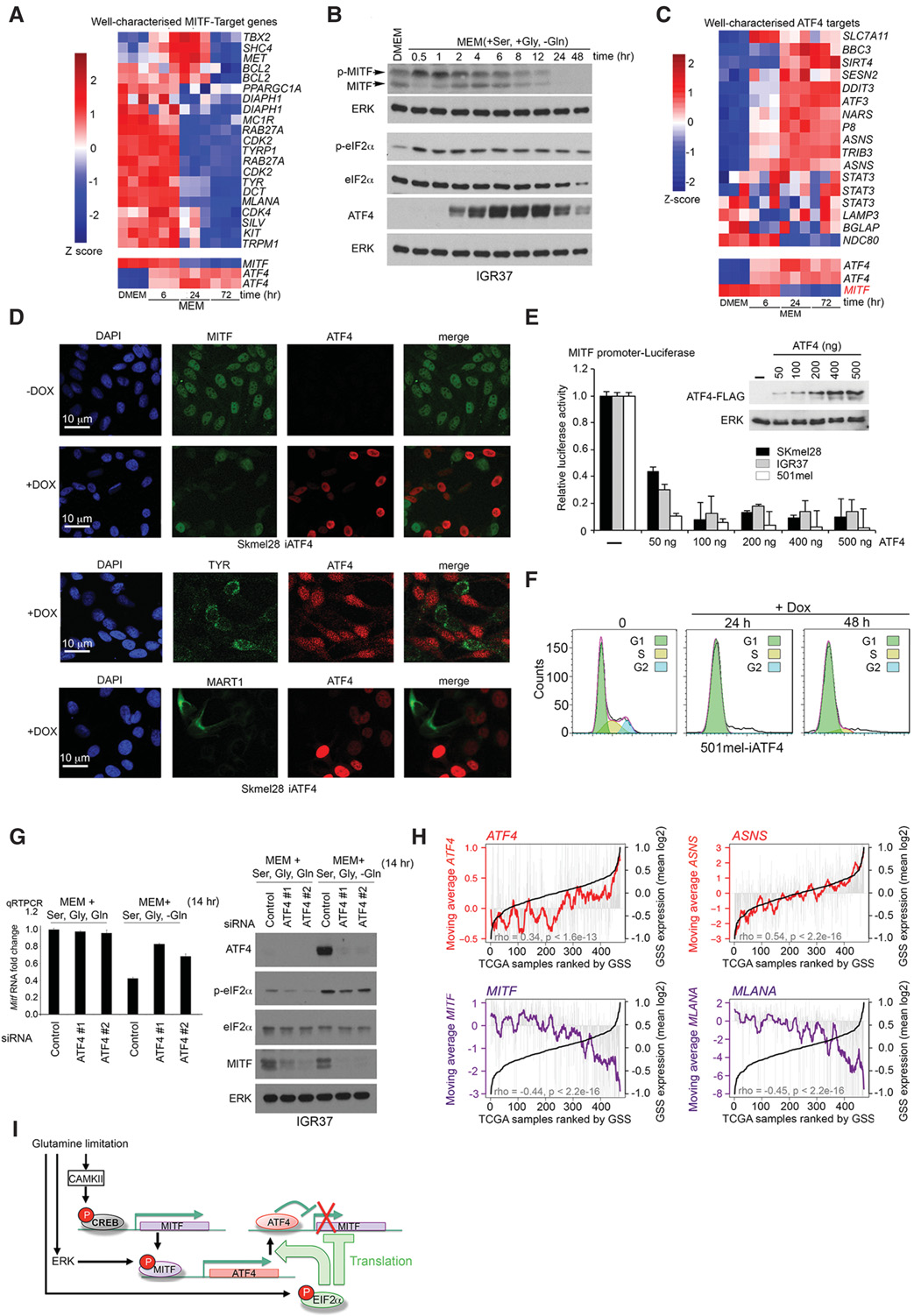
siRNA介导的MITF耗竭最初会驱动侵袭性(Carreira等, 2006),随后引发衰老(Giuliano等,2010),然而矛盾的是,黑色素瘤中存在非衰老MITF阴性细胞。我们认为,如果MITF通过生理机制(如翻译重编程和ATF4)被沉默,则可能绕过siMITF诱导的衰老。与此一致的是,siMITF可诱导衰老,通过衰老相关‐β‐半乳糖苷酶(SA‐β‐gal)活性检测到(图3A,上图;补充图3A),相比之下,通过谷氨酰胺剥夺使MITF表达沉默(图1)并未导致衰老(图3A,下图;<51>补充图S3A)。谷氨酰胺剥夺的细胞S期减少,并在G1期积聚(图3B),但与衰老不同,这种细胞周期阻滞是可逆的,当重新供给谷氨酰胺时,细胞重新进入细胞周期(图3B);即使经过17天的谷氨酰胺饥饿,再供给谷氨酰胺也能逆转MITF及其分化相关靶基因MLANA的丢失(图3C)。值得注意的是,在营养丰富的培养基中诱导 ATF4足以抑制siMITF引起的衰老(图3D)。因此,肿瘤中观察到的MITF低表达非衰老细胞可由促进ATF4翻译增加的信号产生。
ATF4高/MITF低状态不足以驱动侵袭性
GSS与TCGA人类黑色素瘤队列中的两个独立的黑色素瘤侵袭性特征(Hoek等,2006;Verfaillie等,2015)相关 (图3E;补充表S4)。在最近一项基于患者来源的单细胞黑色素瘤RNA测序(RNA‐seq)数据集(Tirosh等, 2016)中,GSS与Verfaillie侵袭特征的相关性更强,这表明与TCGA数据集相关性中的大部分噪声来源于肿瘤内的非黑色素瘤细胞。需要注意的是,GSS与Hoek和Verfaillie特征之间的重叠基因较少,即使去除这些重叠基因,相关性仍可被重现(数据未显示)。GSS还在独立的黑色素瘤细胞系队列(Broad [Lin 等 2008],昆士兰 [Johannessen 等 2013],和杜克 [Augustine 等 2009])中与 Verfaillie侵袭特征相关(补充图S3B,左图),并与它们的侵袭性相关(补充图S3B,右图;Widmer 等,2012)。谷氨酰胺限制促进了侵袭性(图3F),并上调了编码上皮间质转化(EMT)相关转录因子ZEB1、CDH2(N‐钙黏蛋白)和FN1(纤连蛋白1)的信使RNA(补充图S3C)。这一观察结果通过分析谷氨酰胺剥夺细胞的基因芯片数据中 EMT相关基因的表达得到了验证(补充表S1)。ZEB1的上调和SNAI2的下调是晚期黑色素瘤中由EMT相关转录因子介导重编程的标志(Caramel 等,2013)。siRNA介导的 MITF耗竭可触发侵袭(Carreira等,2006),且体内和体外低MITF水平与侵袭性相关。因此,我们预期ATF4介导的MITF抑制会促进从增殖到侵袭性表型的转换。然而令人意外的是,在营养丰富培养基中诱导ATF4(图3F,右图)虽能有效沉默MITF(图2D),却始终未能促进侵袭。由于ATF4高表达/ MITF低表达状态比siRNA耗竭更准确地反映了体内环境,我们重新评估了MITF’s在黑色素瘤进展中的作用。值得注意的是,尽管部分细胞在谷氨酰胺限制条件下死亡,但在iMITF 501mel细胞系中,多西环素介导的 MITF诱导显著增加了细胞死亡(图3G)。这很可能是因为 MITF通过激活CDK表达(Du 等,2004)等方式促进增殖,从而导致一种高营养需求状态,这种状态与营养限制不相容。因此,令人意外的是,ATF4高表达/ MITF低表达状态不足以建立侵袭性表型。这一结果进一步通过检测谷氨酰胺剥夺对不同MITF水平细胞系侵袭性的影响得到证实。在所有情况下,除了MITF阴性的IGR39细胞系外,谷氨酰胺剥夺均导致ATF4表达的瞬时激活,并无论MITF状态如何均增加侵袭性(图3H)。IGR39细胞的不同反应机制尚需进一步研究。
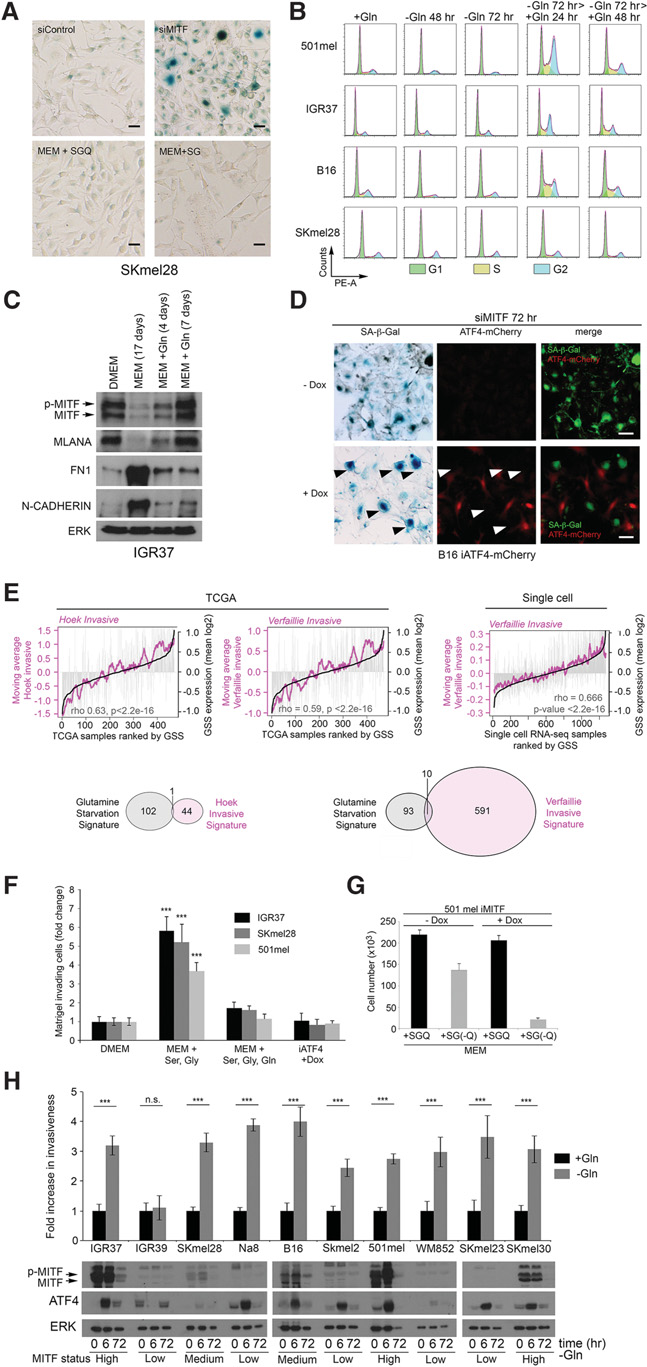
eIF2B抑制驱动侵袭性
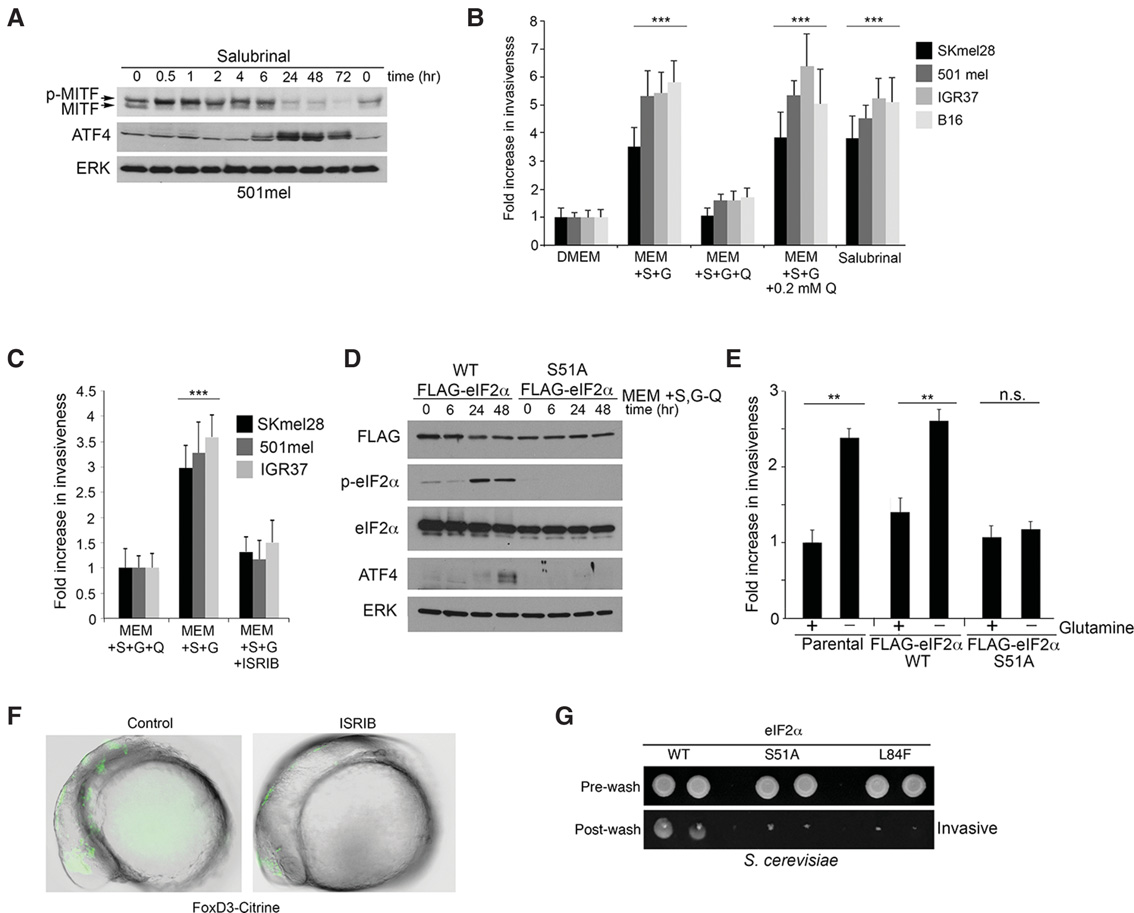
如果如预期的那样,ATF4介导的MITF抑制不足以启动侵袭性,那么什么可以呢?营养限制会增加ATF4表达,但也会通过p‐eIF2α介导的eIF2B抑制引起翻译的全局重编程。然而,当在营养丰富条件下通过多西环素诱导ATF4以抑制MITF时,翻译重编程不会发生。在乳腺癌中,已有报道表明PERK(一种eIF2α激酶)的激活位于上皮间质转化(EMT)的下游(Feng等,2014)。相反,我们的结果表明,侵袭性是eIF2α磷酸化的结果。为了定义p‐eIF2α在侵袭性中的作用,并将其激活与上游信号分离,我们用沙鲁比林处理细胞,沙鲁比林是一种选择性抑制 eIF2α去磷酸化的抑制剂(Boyce等,2005年)。正如预期,沙鲁比林增加了ATF4表达并降低了MITF表达(图4A;补充图S4A),但也下调了E‐钙黏蛋白(补充图S4B),并诱导了侵袭性(图4B)。值得注意的是,ISR抑制剂 ISRIB——一种通过稳定eIF2B二聚体使细胞对eIF2α磷酸化不敏感的药物(Sidrauski等,2015年)——能够阻止由谷氨酰胺限制引起的侵袭性(图4C)。我们使用一组具有不同MITF水平和驱动突变状态的黑色素瘤细胞系发现,沙鲁比林和ISRIB均未引起显著的细胞死亡(补充图S4C),并且尽管NRAS突变细胞系在谷氨酰胺剥夺时似乎对死亡的敏感性较低,但仍需更广泛的分析来确定其是否具有显著性。
为了验证eIF2α磷酸化在侵袭中的作用,我们在501mel细胞中稳定表达了Flag标签的eIF2α野生型以及一种起显性失活作用的S51A突变体,并检测它们对谷氨酰胺限制的反应。结果表明,表达外源性野生型eIF2α的细胞诱导了 ATF4(图4D)并表现出侵袭性(图4E),而表达显性失活eIF2α S51A突变体均未能实现上述作用。综上所述,这些数据明确表明eIF2B抑制是黑色素瘤侵袭性的关键驱动因素。值得注意的是,该通路也可被奈非那韦激活(补充图S4D),奈非那韦是一种最近被提议用于黑色素瘤治疗的药物(史密斯等人,2016年),这意味着在体内暴露于亚致死浓度奈非那韦的细胞可能更具侵袭性。
由于黑色素瘤起源于具有迁移性神经嵴发育起源的黑色素细胞,我们探究了ISRIB是否也会阻断神经嵴的迁移。利用荧光标记神经嵴细胞的斑马鱼模型,在未经处理的胚胎中可明显观察到神经嵴细胞的迁移,而在ISRIB处理的鱼类中,神经嵴细胞则滞留在中线位置(图4F)。尽管由于缺乏相应工具,我们未能在鱼类神经嵴中确认ISRIB对 ATF4、MITF和eIF2α的影响,但这一结果仍表明,由神经嵴发出的信号所施加的翻译重编程汇聚于eIF2B抑制,实际上劫持了饥饿反应以促进迁移。
为了提供遗传证据,证明翻译重编程作为进化保守驱动因素在侵袭行为中的作用,我们还测试了eIF2B的抑制是否为酵母侵袭所必需。在营养胁迫条件下,出芽酵母酿酒酵母会形成侵袭性链状结构,表现出丝状生长表型(吉梅诺等人,1992年)。将携带野生型eIF2α基因、S51A突变体或L84F突变体(允许eIF2α‐S51磷酸化但阻止与eIF2B相互作用)的酵母在琼脂上培养。用水冲洗后,仅有侵入基质的细胞被保留下来。该实验表明,具有野生型eIF2α的细胞具有侵袭性,而携带eIF2α S51A或L84F突变的酵母则没有这种现象(图4G)。这一遗传证据有力地支持了我们的观点,即迁移/侵袭性是针对eIF2B抑制及随之发生的翻译重编程的一种进化保守反应。此外,由于酵母没有MITF基因,该结果再次强调了我们之前的观察(图3F,H),即侵袭性可以与 MITF表达的影响相分离。
肿瘤坏死因子 α(TNFα)介导翻译重编程、ATF4表达和 MITF抑制
鉴于酵母、神经嵴和黑色素瘤中的侵袭性是通过抑制 eIF2B来实现的,我们预测其他肿瘤内微环境信号会劫持这种进化保守的饥饿反应,即使在营养丰富条件下也通过相同机制诱导侵袭性。来源于浸润性免疫细胞的炎症性细胞因子在肿瘤进化中起关键作用。其中较为重要的一种是 TNFα。Smith等人(2014)报道称,TNFα通过上调 MITF促进黑色素瘤生长和治疗抗性,这应会促进分化。相反,其他研究表明TNFα下调MITF(Landsberg 等人 2012;Konieczkowski等,2014;Riesenberg等人2015),减少分化,并促进慢循环表型(Ostyn等人,2014),这是 MITF低表达细胞的特征(Cheli等人 2011b)。值得注意的是,最近描述的一组黑色素瘤TNFα反应基因( Riesenberg等人 2015)与TCGA黑色素瘤队列以及 Tirosh等人(2016)提供的单细胞RNA‐seq患者来源黑色素瘤数据中的GSS评分呈正相关(图5A)。两个特征之间的重叠很小,即使去除重叠基因,相关性仍可重现(数据未显示)。这表明谷氨酰胺饥饿和TNFα的反应汇聚于一种共同的黑色素瘤表型。在培养条件下,尽管不同细胞系间的反应存在差异,TNFα引发的反应与谷氨酰胺限制下观察到的反应极为相似(图5B);任何MITF的增加(如 Smith等人(2014)所观察到的)都是短暂的,而在所有细胞系中,TNFα最终促进了ATF4表达(提示p‐eIF2α介导的翻译重编程)和侵袭性(图5C),这与TNFα驱动 MITF低表型和去分化的现象一致。
为了进一步探究炎症、侵袭性和谷氨酰胺饥饿基因表达程序在体内的汇聚,我们使用了来自炎症介导的黑色素瘤去分化小鼠模型的基因表达数据(图5D,左; Landsberg 等人 2012)。在此模型中,肿瘤最初对采用转基因细胞毒性pmel‐1 T细胞的过继细胞转移疗法有反应,该疗法靶向黑色素瘤分化抗原gp100(也称为Pmel)。然而,T细胞和肿瘤浸润性髓系免疫细胞释放的TNFα和其他细胞因子在体内诱导黑色素瘤细胞去分化,导致脱色素、 gp100抗原下调以及逃避免疫疗法pmel‐1 T细胞。所使用的HCmel3小鼠黑色素瘤细胞系也对谷氨‐酰胺剥夺和/或TNFα暴露通过下调MITF实现(图5D,右)。对由炎症驱动的去分化复发性黑色素瘤基因表达谱进行基因集富集分析(GSEA)(图5E),结果显示其与TNFα应答基因以及Hoek和Verfaillie侵袭性基因集呈显著正相关,这与TNFα介导的去分化驱动促侵袭表型转换一致。值得注意的是,体内基因表达数据还显示与GSS存在显著正相关,且复发性肿瘤中Hoek和Verfaillie增殖性基因表达特征明显减少(图5E;补充表S4)。此外,Mitf及其靶基因 Mlana在复发性肿瘤中显著下调,而Atf4和Asns则显著上调(图5F)。尽管我们并未证明翻译重编程与体内pmel过继性T细胞治疗耐药性之间存在因果关系,但总体而言,这些数据表明与过继性T细胞疗法相关的体内炎症介导的去分化表型转换最终是通过一种类似于谷氨酰胺剥夺所观察到的应激反应而产生的。
ATF4驱动MITF低/AXL高药物耐药特征
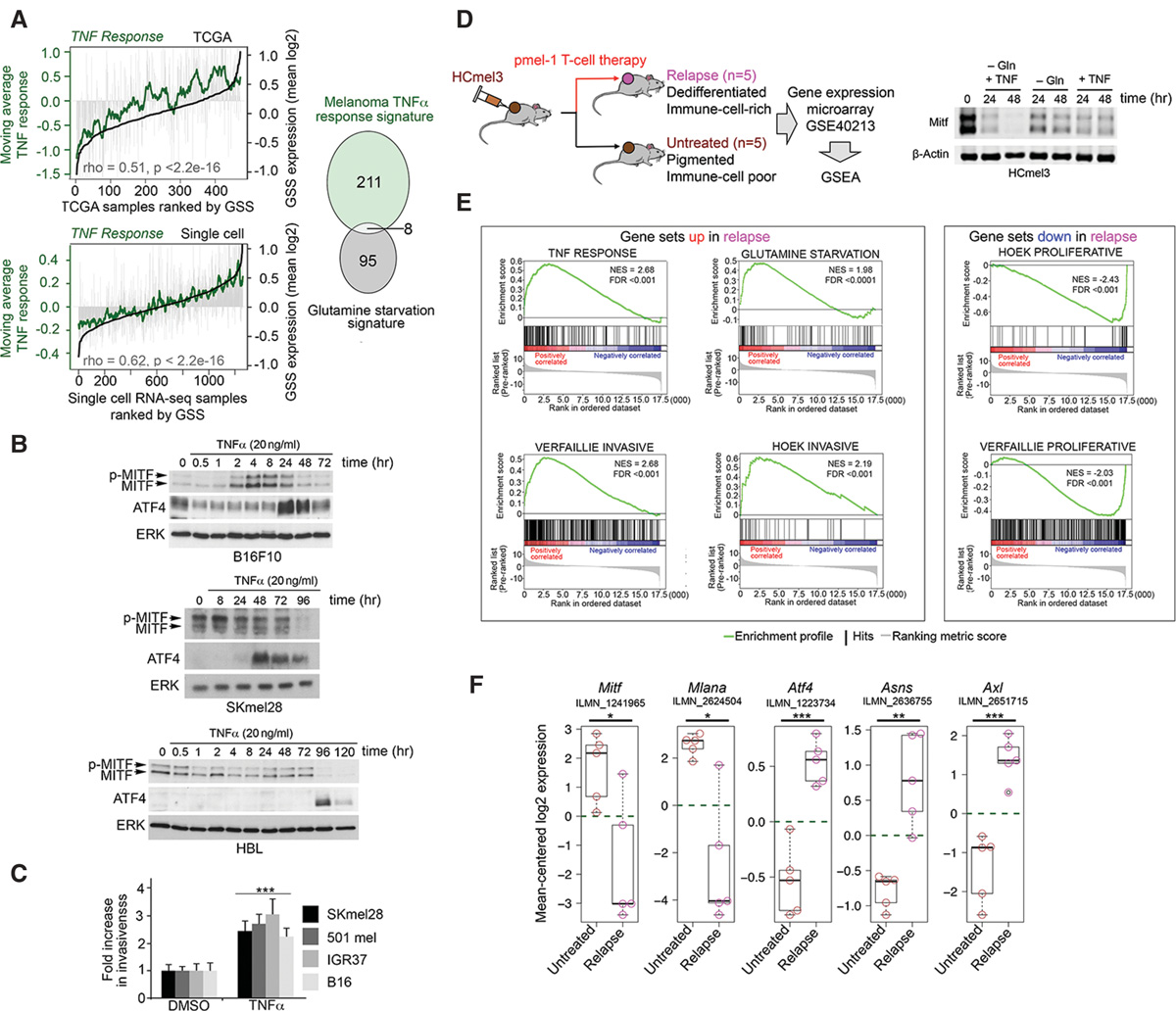
引人注目的是,复发的小鼠肿瘤也表现出AXL受体酪氨酸激酶的显著上调(图5F);MITF低/AXL高表型(补充图 S4E)是人类黑色素瘤中BRAF抑制剂耐药性的明确标志 (Konieczkowski等,2014;Muller等,2014;Dugo等, 2015;Tirosh等,2016),并且已被证实与头颈部癌中抗 PI3K治疗的耐药性相关(Elkabets等,2015)。AXL表达上调的具体机制尚不明确。然而,在TCGA黑色素瘤样本中, AXL水平与GSS评分呈正相关(图6A,左图),而100基因 AXL程序特征(补充表S4;Tirosh等,2016)在 Tiroshet等(2016)提供的单细胞黑色素瘤基因表达谱中也与GSS高度相关(图6A,右图)。值得注意的是,谷氨酰胺耗竭(图6B,C)或在营养丰富培养基中通过多西环素诱导ATF4表达(图6D;补充图S4F)均可导致MITF低/ AXL高状态,提示驱动肿瘤内ATF4蛋白表达的应激可能启动了所观察到的MITF低/AXL高表型。
eIF2B抑制与IPRES(先天性抗PD‐1耐药)基因表达特征相关
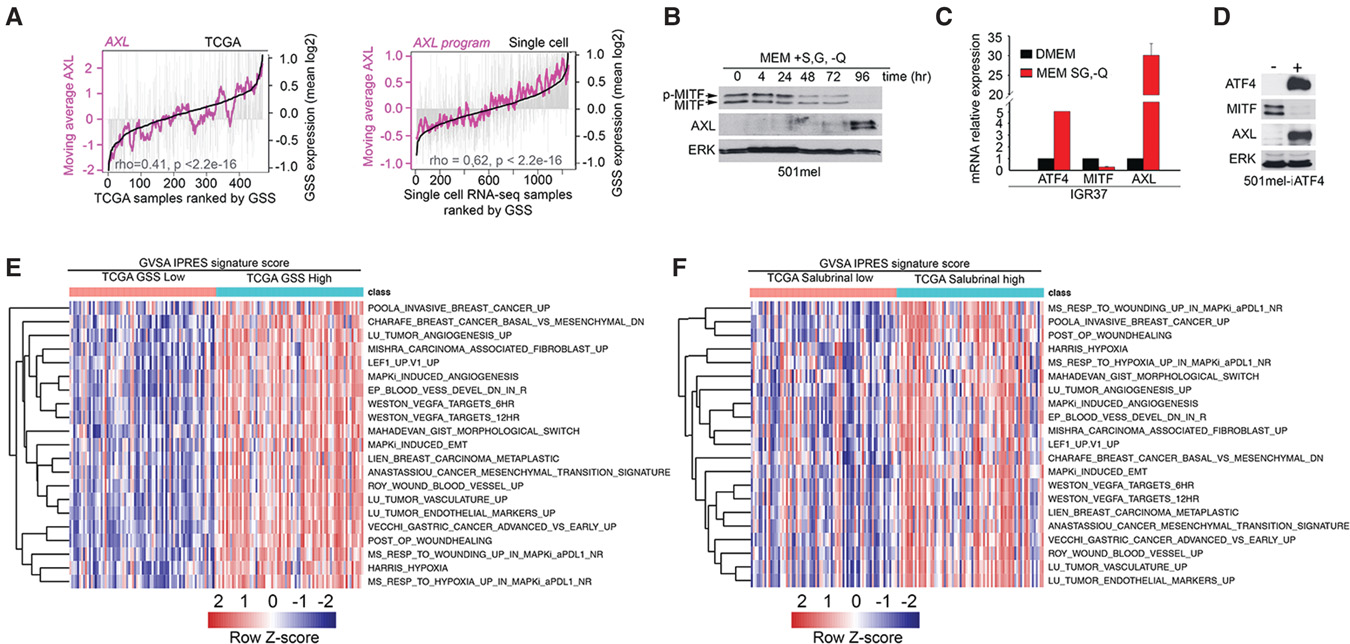
T细胞相关PD‐1与其配体PD‐L1(PD‐1)之间的相互作用会抑制效应T细胞功能,从而保护黑色素瘤免受免疫排斥。因此,阻断PD‐1/PD‐L1轴可带来显著的治疗益处( Wolchok等,2013)。然而,对抗PD‐1治疗无响应的情况与一种特征性基因表达模式相关,该模式还包括AXL表达升高,称为IPRES特征(Hugo 等人 2016)。尽管IPRES特征存在于大部分无应答肿瘤中,但其形成机制尚不清楚。由于AXL表达与抗 PD‐1治疗反应不佳相关(Hugo 等人 2016)且可被ATF4诱导,我们探讨了IPRES基因表达特征是否与TCGA黑色素瘤队列中的GSS评分相关。通过基因集变异分析(GSVA)比较按GSS评分排序的最高和最低75个黑色素瘤中的 IPRES特征,结果显示了极强的相关性(图6E)。重要的是,我们使用经24小时沙鲁比林处理细胞所获得的基因表达特征也观察到了类似的富集情况(补充表S4、S5),通过促进eIF2α磷酸化来抑制eIF2B。因此,翻译重编程不仅与侵袭性、去分化和对过继T细胞治疗的耐药性相关,还可施加一种基因表达程序,从而预测抗PD‐1免疫治疗的不良反应。
翻译重编程促进肿瘤定植
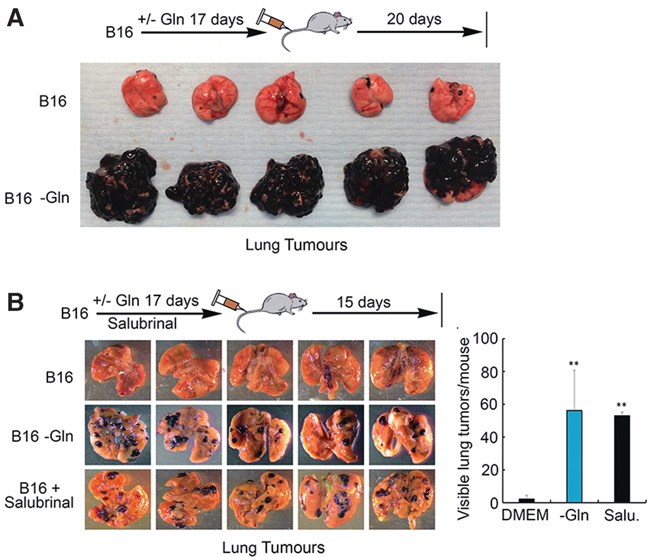
罕见的、随机发生的慢周期MITF低表达细胞具有增强的肿瘤起始能力(Cheli等人 2011b)。然而,这些细胞在体内如何产生尚不清楚。由于谷氨酰胺剥夺可产生MITF低表达细胞,因此翻译重编程可能控制肿瘤启动能力。我们因此将B16黑色素瘤细胞注射到免疫功能健全的C57/Bl6小鼠的尾静脉中,并评估肺部肿瘤形成情况,该实验可测量细胞在循环系统中的存活能力以及定量评估肿瘤启动能力。注射20天后,肺部可见少量小肿瘤(图7A),这与罕见的MITF低表达肿瘤启动细胞的存在一致(Cheli等人 2011b)。相比之下,注射前经谷氨酰胺剥夺处理的B16细胞导致严重的肿瘤负荷。由于 eIF2B抑制足以增加侵袭性,我们在尾静脉注射实验中比较了沙鲁比林与谷氨酰胺剥夺的作用。注射15天后对小鼠进行检查,此时未经处理的B16细胞形成的肿瘤几乎不可见 (图7B)。相比之下,注射谷氨酰胺缺乏的或经沙鲁比林处理的细胞的小鼠肺部可见大量色素性肿瘤,表明在此实验中eIF2B抑制可增强肿瘤起始能力。
讨论
鉴于MITF在整合黑色素瘤生物学众多方面的作用,以及 MITF低表达状态的事实与耐药性相关,了解肿瘤微环境如何影响MITF表达对于开发有效的抗黑色素瘤疗法至关重要。本文揭示了综合应激反应(ISR)与由MITF调控的黑色素瘤生物学背后的基因表达程序之间一个关键且此前未被察觉的联系:微环境信号(包括谷氨酰胺限制和TNFα)汇聚并抑制eIF2B,从而阻断MITF的翻译以及ATF4表达,并直接抑制MITF转录。尽管我们未研究ATF4介导MITF抑制的分子机制,但其可能通过与MITF启动子激活因子CREB竞争结合相似的 DNA结合基序而将其置换,或通过招募抑制性共因子实现,后者已在Apelin基因启动子上被观察到(Jeong 等人 2014)。
ATF4和翻译重编程在过继性T细胞治疗耐药性中的作用具有临床相关性。抗PD1或PD‐L1治疗在相当比例的患者中无效,耐药性问题日益突出,目前仍在持续努力改进过继性T细胞治疗或开发通过T细胞激活发挥作用的抗黑色素瘤疫苗。识别潜在的耐药机制,例如通过ATF4介导的炎症抑制分化以及翻译介导的MITF抑制,为靶向翻译重编程并恢复对T细胞介导疗法的敏感性提供了机会。此外,虽然谷氨酰胺剥夺和沙鲁比林处理并未显示出对免疫系统与黑色素瘤细胞相互作用的直接影响,但两者均产生一种基因表达特征,可用于预测已建立的免疫治疗耐药性特征(IPRES)(Hugo 等人 2016)的存在。这表明翻译重编程可能在响应微环境信号时调节黑色素瘤细胞与免疫系统之间相互作用中发挥关键作用,尽管仍需进一步研究来明确其实现机制。重要的是,ATF4表达还可驱动此前未被解释的MITF低表达/AXL高表达基因表达程序( Konieczkowski等,2014;Muller等,2014;Dugo等, 2015),这是BRAF抑制剂耐药性的标志。
除了与耐药性相关外,MITF低表达细胞还具有侵袭性。以往研究的一个结论是,侵袭与增殖呈负相关(Hoek 等, 2006),并且MITF在某种程度上抑制了侵袭性。然而,此前尚未明确为何侵袭与增殖呈负相关,以及MITF如何可能抑制侵袭。我们的研究结果提供了一种解释:响应包括营养缺乏、炎症及其他上游应激在内的信号,eIF2B受到抑制,从而使细胞启动双臂适应性反应:首先,通过减少为增殖所必需的大部分翻译,eIF2B的抑制降低了营养需求;其次,细胞通过MITF增加ATF4转录并增强ATF4的翻译,从而驱动氨基酸摄取和自噬以增加营养供应。此外,通过翻译重编程介导的侵袭性可进一步提高营养供应,使细胞得以逃离应激环境并寻找新的营养来源。因此,增殖与侵袭性之间存在负相关关系,其中全局翻译受到抑制。
我们还表明,在营养丰富的条件下,ATF4的诱导可减少MITF mRNA的表达,从而产生一种在肿瘤中观察到的 ATF4高/MITF低非衰老状态,但令人惊讶的是,这种状态不足以促进侵袭。结合我们观察到的在谷氨酰胺受限条件下维持MITF表达与存活不相容的结果,我们认为MITF促进增殖(一种高营养需求状态)的能力与营养限制不相容;真正驱动侵袭性的关键因素是通过抑制eIF2B介导的翻译重编程,而不是MITF表达的降低或ATF4的升高;但必须降低MITF的表达才能允许细胞存活。这解释了为何增加 MITF表达的治疗策略(例如使用甲氨蝶呤)能够非常有效地防止转移扩散(Saez‐Ayala等人,2013年)。更重要的是,我们发现翻译重编程可以抑制MITF,这一发现对那些有意(Cerezo等人,2016年)或无意中上调黑色素瘤内质网应激和ATF4的药物的治疗应用具有重要意义。奈非那韦最近在一项药物再利用筛选中被鉴定为一种可下调MITF的黑色素瘤治疗药物(史密斯等人,2016年),已知其可抑制一种eIF2α磷酸酶(De Gassart等人,2016年)。尽管据报道其通过抑制PAX3来下调MITF,但我们表明奈非那韦也可在黑色素瘤中促进翻译重编程和ATF4表达。我们认为,尽管通过应激诱导的ATF4翻译来下调MITF在某些情况下可能具有治疗优势(例如当细胞对MITF表达的动态变化敏感时),但存在一种风险:通过促进翻译重编程的药物来抑制MITF可能会在体内许多细胞可能遇到的亚致死剂量下增加转移扩散。
尽管我们在此主要关注通过ATF4和eIF2α的磷酸化产生MITF低表达的非衰老黑色素瘤细胞,但显然其他因素也可以在转录水平上沉默MITF。这些因素包括BRN2( POU3f2),其在肿瘤中的表达与MITF相互排斥( Goodall 等,2008),以及在缺氧条件下被激活的 BHLHB2(Cheli 等,2011a)。虽然BRN2介导的MITF抑制可诱导侵袭,但尚不清楚它是否通过增加eIF2α的磷酸化来促进侵袭。相比之下,低氧水平确实会导致内质网应激和p‐eIF2α升高(Koumenis 等,2002),这可能有助于在缺氧条件下观察到的侵袭性(Cheli 等, 2011a)。
先前在黑色素瘤中的研究已经证实,矛盾的是,在培养中罕见的、随机发生的缓慢循环细胞比快速增殖细胞具有更强的肿瘤启动能力(罗施等人 2010;切利等人 2011b)。我们的结果表明,通过沙鲁比林处理抑制 eIF2B 后细胞的肿瘤启动能力增强,可能反映了由翻译转换直接驱动的四种适应性变化的综合作用:第一,由于整体翻译水平降低导致营养需求减少,从而产生慢周期细胞,这些细胞在体内遇到的低营养环境中比高需求的增殖细胞具有更强的存活能力。第二,细胞表面黏附分子的变化将使细胞能够黏附于适合存活的微环境。第三,ATF4 可促进对失巢凋亡的耐药性并抑制氧化应激(德伊等人 2015),而应对氧化应激是成功建立转移灶的关键决定因素(皮斯昆诺娃等人 2015)。第四,我们发现沙鲁比林或谷氨酰胺限制可驱动一种此前无法解释的与抗PD‐1免疫治疗无响应相关的基因表达程序(IPRES),提示翻译重编程可能引发一种免疫豁免状态。因此,响应翻译重编程而离开原发肿瘤的细胞,对迁移过程中伴随的各种应激具有极强的适应能力。
总之,我们的研究揭示了一条此前未被充分认识的翻译重编程/ATF4–MITF轴,该轴整合多种微环境信号,导致MITF受到抑制和黑色素瘤去分化,并揭示了位于ATF4上游的翻译重编程驱动了与治疗抗性和转移扩散相关的基因表达程序。

















 1025
1025

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









