



基于窄带隙电子受体的高效聚合物太阳能电池的设计以实现优异的短路电流密度
摘要
为了拓展吸收范围并增强稠环电子受体(FREAs)的电子迁移率,我们基于4,9‐二氢噻吩并[3’ ,2’:4,5]环戊二噻吩并[1,2‐b]噻吩并[2’‘,3’‘:3’,4’]环戊二噻吩并[1’,2’:4,5]噻吩并‐d]噻吩(DTCTT)设计并合成了新的FREAs(TTIC和TTIC‐M)。1,1‐二氰亚甲基‐3‐茚酮(IC)。DTCTT单元具有强的给电子能力,可通过增强的内电荷转移(ICT)效应拓宽所得受体分子的吸收。稠环结构上的硫原子可通过分子间非共价S‐S相互作用增强电子传输。此外,在端基IC单元上引入甲基可提高非富勒烯受体(FREAs)的LUMO能级,从而获得更高的开路电压(Voc)
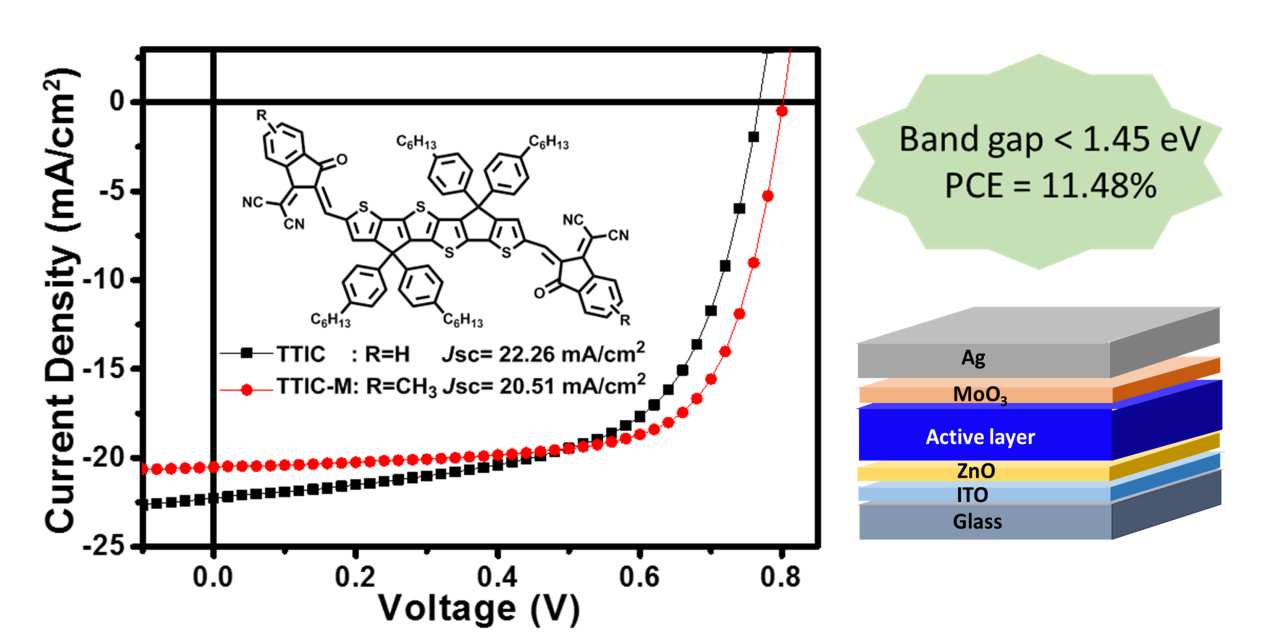
正如预期,TTIC和TTIC‐M表现出宽广的吸收,其吸收边延伸至近红外区域(900纳米),TTIC和TTIC‐M的光学带隙分别为1.40和1.44电子伏特。基于PBDB‐T:TTIC的器件实现了10.61%的光电转换效率,短路电流密度高达22.26 mA cm⁻²,开路电压为0.77 V,填充因子为0.62。更值得注意的是,基于PBDB‐T:TTIC‐M的器件提供了更高的开路电压0.80 V,光电转换效率达到11.48%,短路电流密度为20.51 mA cm⁻²,填充因子为0.70。11.48%是迄今为止基于窄带隙非富勒烯受体(<1.45电子伏特)的聚合物太阳能电池所取得的最佳性能。
1. 引言
体异质结聚合物太阳能电池(PSCs)具有加工成本低、半透明性、柔性和轻质等显著特点,被认为是极具前景的下一代太阳能电池。[1‐4]通常,体异质结PSCs由一种可溶液加工的p型有机半导体作为供体和一种富勒烯衍生物作为电子受体组成。在过去几十年中,PSCs主要采用富勒烯衍生物(如PC61BM、PC71BM和ICBA)作为电子受体。研究人员在开发新型供体材料、优化器件结构以及调控共混薄膜形貌方面投入了大量努力,近年来已实现PSCs的功率转换效率(PCE)超过11%。[5]然而,富勒烯受体存在一些固有缺陷,例如在可见光区域吸收较弱,甚至在近红外(NIR)区域无吸收、能级可调性有限、形貌稳定性差以及较大的能量损失,限制了它们未来的实际应用。与富勒烯受体相比,非富勒烯电子受体具有显著优势,例如具有增强的吸收系数的宽吸收、可调的分子能级、良好的器件稳定性、易于合成与纯化以及潜在的低成本生产。[6‐11]由于上述原因,通过分子结构优化,非富勒烯电子受体得到了迅速发展和进步,光电转换效率已超过13%。[12]
最近,稠环电子受体(FREAs)引起了广泛关注。[13]大量FREAs,如ITIC、[14] ITIC‐Th、[15] IEIC、[16]IDTBR、[17]和IDIC,[13]它们包含IDT或IDTT给体单元以及1,1‐二氰亚甲基‐3‐茚酮(IC)受体单元,已被报道并实现了令人印象深刻的光电转换效率。这些FREAs的分子结构如图1所示。尽管这些FREAs与多种聚合物给体具有良好兼容性,但只有少数能够提供超过10%的高光电转换效率和高的短路电流密度(Jsc)(>20 mA cm⁻²)。[12]一个可能的原因是这些受体仅在300至800 nm范围内吸收光。[18]另一个可能的原因是基于这些受体的共混薄膜通常表现出较低的电子迁移率。[19]

为了拓展吸收范围并增强非富勒烯受体(FREAs)的电子迁移率,我们设计并合成了一类基于4,9‐二氢噻吩并[3’ ,2’:4,5]环戊二噻吩并[1,2‐b]噻吩并[2’‘,3’‘:3’,4’]环戊二噻吩并[1’,2’:4,5]噻吩并‐d]噻吩(DTCTT)和IC的新型非富勒烯受体(TTIC和TTIC‐M)。DTCTT单元比IDT和IDTT具有更强的给电子能力,可通过增强的内电荷转移(ICT)效应拓宽所得受体分子的吸收。稠环结构上的硫原子可通过分子间非共价S‐S相互作用增强电子传输。此外,在端基IC单元上引入甲基可提高FREAs的LUMO能级,从而获得更高的开路电压(Voc)。
这两种新型非富勒烯受体(TTIC和TTIC‐M)表现出宽广的吸收,吸收边延伸至近红外区域(900纳米),对应的光学带隙分别为TTIC的1.40电子伏特和TTIC‐M的1.44电子伏特。当与宽带隙聚合物给体PBDB‐T共混时,基于TTIC的器件实现了10.61%的光电转换效率,并具有出色的Jsc(22.26毫安/平方厘米)。随着预期的是,由于LUMO能级升高,基于TTIC‐M的器件表现出更高的Voc。实现了11.5%的高PCE,其中Jsc为20.51 mA cm⁻²,Voc为0.80 V,FF为0.70。据我们所知,11.48%的PCE是迄今为止基于小带隙(<1.45 eV)非富勒烯受体的钙钛矿太阳能电池的最佳结果。
2. 实验部分
2.1. 材料合成与表征仪器
所有试剂和化学品均购自商业供应商,除特别说明外,未经进一步纯化即使用。溶剂在使用前按标准方法蒸馏。除非另有说明,所有反应均在氮气气氛下进行,并通过硅胶薄层色谱(TLC)监测。柱层析使用硅胶(200‐300目)进行。
PBDB‐T:poly[(2,6‐(4,8‐双(5‐(2‐乙基己基)噻吩‐2‐基)‐苯并[1,2‐b:4,5‐b’]二噻吩))‐alt‐(5,5‐(1’,3’‐二‐2‐噻吩基‐5’,7’‐双(2‐乙基己基)苯并[1’,2’‐c:4’,5’‐c’]二噻吩‐4,8‐二酮))]根据文献[23]合成。
¹H和¹³C核磁共振谱在布鲁克Advance 500 MHz仪器上使用CDCl₃作为溶剂进行测定。热重分析(TGA)在梅特勒‐托利多TGA/DSC 1/1100SF仪器上进行。样品的热稳定性通过在氮气氛围下以10 °C/min的升温速率测量其重量损失来确定。差示扫描量热法(DSC)测量在珀金埃尔默Diamond DSC仪器上进行。紫外‐可见(UV‐Vis)吸收光谱在珀金埃尔默Lambda 750型紫外‐可见分光光度计上测定。
元素分析在Flash EA 1112分析仪上进行。循环伏安图(CV)测量在ModuLab XM电化学分析仪(英国Solartron Analytical有限公司)上进行,采用三电极电化学池,在0.1 M四(正丁基)铵六氟磷酸盐(Bu₄NPF₆)溶液中,扫描速度为100 mV/s,室温下氮气氛围中进行。对电极和参比电极分别使用铂丝和Ag/AgCl。透射电子显微镜(TEM)在JOEL JEM‐2100透射电镜上以200 kV进行观测。原子力显微镜(AFM)测量在Digital Instrument Multimode Nanoscope IIIA仪器上以轻敲模式进行。共混薄膜的厚度由Dektak 6M表面轮廓仪测定。
2.2 太阳能电池的制备与表征
光伏电池采用以下步骤制备,器件结构为ITO/ZnO(30 纳米)/活性层(85 纳米)/MoO₃(8.5 纳米)/Ag(100 纳米)。氧化铟锡(ITO)玻璃在超声波清洗槽中依次用水、丙酮、异丙醇和水清洗,每次20分钟。基底在加热板上于160 °C加热15分钟以烘干,随后进行20分钟氧气等离子体处理。ZnO前驱体通过将乙酸锌二水合物(Aldrich,99.9%,1克)和乙醇胺(Aldrich,99.5%,0.28克)溶解于2‐甲氧基乙醇(Aldrich,99.8%,10毫升)中,在剧烈搅拌下反应12小时以完成水解反应而制得。将ZnO前驱体在3500转/分钟的转速下旋涂于基底上,持续15秒。然后将覆盖有ZnO前驱体的基底在200 °C加热20分钟。
将聚合物给体与非富勒烯受体的混合溶液(聚合物浓度为4 毫克/毫升)在氧化锌层上以1200 转/分钟的速度旋涂。随后在活性层上蒸镀8.5 纳米的MoO₃层和100 纳米的银层。每个电池的有效面积为0.04 cm²。电流‐电压特性在室温下使用吉时利2400源表记录,测试采用AM1.5G AAA级太阳光模拟器(型号SS‐F53A,台湾友力科技有限公司),光强为100 mW/cm²,白光强度通过标准单晶硅太阳能电池进行校准。外量子效率(EQE)测量在空气中未封装条件下进行。
2.3 合成
化合物2 。在氮气保护下,将化合物1(524 毫克,0.54 毫摩尔)的四氢呋喃(30 毫升)溶液于‐78 °C缓慢加入1.35 毫升(3.24 毫摩尔)2.4 M 正丁基锂的己烷溶液。混合物在0 °C保持一小时,然后在‐78 °C加入N,N‐二甲基甲酰胺(1 毫升)。混合物搅拌过夜至恢复室温。加入饱和氯化钠溶液,分离有机层。水层用CH₂Cl₂萃取三次;合并的有机层用无水MgSO₄干燥并蒸干。残余物通过硅胶柱柱层析纯化,以石油醚和CH₂Cl₂(3:2 体积比)洗脱,得到化合物2为橙色固体(167 mg,30%)。熔点(Mp)218 °C。¹H NMR (500 MHz, CDCl₃),(ppm):9.80 (s, 2H),7.68 (s, 2H),7.12 (m, 16H),2.56 (m, 8H),1.57 (m, 8H),1.29 (m, 24 H),0.86 (t, J= 6.7 Hz, 12H)。¹³C NMR (125 MHz, CDCl₃),(ppm):182.45,157.99,152.38,146.96,144.28,142.62,138.28,138.16,137.64,131.64,128.88,127.56,62.32,35.58,31.69,31.25,29.10,22.59,14.09。IR (KBr, cm⁻¹):2954,2924,2853,1655,1498,1395,1319,1222,1141。元素分析计算值 C₆₆H₇₂O₂S₄:C, 77.30;H, 7.08。实测值:C, 77.21;H, 7.07%。
TTIC通过2和IC(2‐(3‐氧代‐2,3‐二氢‐1H‐茚‐1‐亚基)丙二腈)的克脑文盖尔缩合反应合成。将2(105毫克,0.10毫摩尔)和IC(300毫克,1.54毫摩尔)的氯仿溶液(30毫升)置于氮气氛围下,加入吡啶(2毫升)。混合物在氮气氛围下加热并搅拌过夜,温度为65 °C。反应完成后冷却,浓缩反应液,再经硅胶柱层析纯化,以石油醚和CH₂Cl₂(3:2 体积比)洗脱,得到深蓝色固体TTIC(72毫克,51%收率)。熔点(Mp)为312 °C。¹H NMR(500 MHz,CDCl₃),ppm:8.86(s,2H),8.67(m,2H),7.90(m,2H),7.72(m,6H),7.14(m,16H),2.58(m,8H),1.59(m,8H),1.32(m,24H),0.86(t,J=6.5 赫兹,12H)。¹³C NMR(125 MHz,CDCl₃),ppm:188.47, 160.28, 159.56, 155.42, 154.49, 142.88, 139.96, 139.72, 138.67, 137.77, 136.79, 134.99, 134.26, 129.02, 127.56, 125.22, 123.62, 114.85, 114.83, 68.34, 62.21, 35.57, 31.67, 31.22, 29.07, 22.56, 14.07。IR(KBr,cm⁻¹):2953, 2924, 2853, 2217, 1697, 1537, 1478, 1398, 1284, 1245, 1128, 981。元素分析计算 C₉₀H₈₀N₄O₂S₄:C, 78.45;H, 5.85;N, 4.07。实测值:C, 78.46;H, 5.83;N, 4.08%。
TTIC‐M 通过化合物 2 与 IC‐M 的克脑文盖尔缩合反应制备得到(其中 IC‐M 为 2‐(5‐甲基‐3‐氧代‐2,3‐二氢‐1H‐茚‐1‐亚基)丙二腈 或 2‐(6‐甲基‐3‐氧代‐2,3‐二氢‐1H‐茚‐1‐亚基)丙二腈)。将化合物 2(150 毫克,0.15 毫摩尔)、IC‐M(150 毫克,0.72 毫摩尔)和氯仿(30 毫升)的混合物在加入吡啶(2 毫升)前后均进行脱气处理,并用氮气置换。混合物在氮气氛围下于 65 °C 加热搅拌 12小时。反应完成后冷却,浓缩反应液,再经硅胶柱层析纯化,以石油醚和 CH₂Cl₂(3:2 体积比)洗脱,得到深蓝色固体 TTIC‐M(40 毫克,产率 19%)。熔点(Mp)323 °C。¹H NMR(500 MHz,CDCl₃),(ppm):8.85(s,2H),8.57(d,J=10赫兹,0.6H),8.49(s,1.4H),7.81(m,1.4H),7.71(s,2.6H),7.56(m,2H),7.16(m,16H),2.59(m,14H),1.61(m,8H),1.31(m,24H),0.89(t,J=7.5 赫兹,12H)。¹³C NMR(125 MHz,CDCl₃),(ppm):188.26, 160.45, 159.37, 154.31, 146.59, 142.86, 142.77, 140.40, 139.59, 138.45, 137.87, 137.57, 135.29, 134.70, 129.02, 127.59, 125.61, 125.19, 123.55, 121.95, 114.99, 114.92, 62.23, 35.60, 31.69, 31.25, 29.10, 22.59, 14.10。IR(KBr,cm⁻¹):2953, 2925, 2854, 2217, 1697, 1537, 1474, 1399, 1367, 1284, 1250, 1127, 986, 910。元素分析计算 C₉₂H₈₄N₄O₂S₄:C, 78.59;H, 6.02;N, 3.99。实测值:C, 78.45;H, 6.03;N,3.99%。
3. 结果与讨论
3.1 材料合成与表征
两种新型受体(TTIC和TTIC‐M)以及聚合物PBDB‐T的化学结构如图3所示。
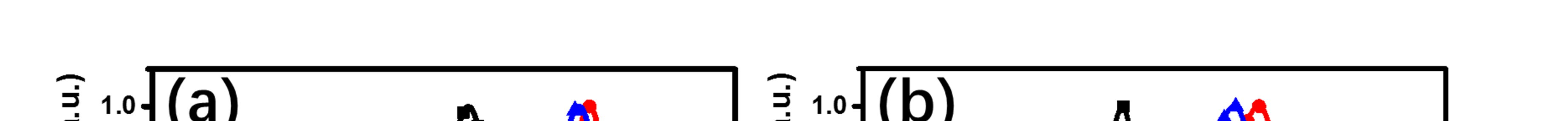
TTIC和TTIC‐M通过克脑文盖尔缩合反应合成,产率分别为51%和19%。详细的合成步骤见实验部分。这两种受体在室温下可完全溶于二氯甲烷、氯仿、四氢呋喃、氯苯和邻二氯苯(DCB)等常见有机溶剂。在氮气氛围下通过热重分析(TGA)研究了TTIC和TTIC‐M的热性能(图S1a),结果表明它们具有良好的热稳定性,其分解温度(以5%质量损失计)分别为349和359 °C。没有明显的玻璃化转变温度在80至350°C范围内通过差示扫描量热法(DSC)测量观察到的转变(图S1b)。
3.2. 光学性质
研究了TTIC和TTIC‐M在溶液中及薄膜状态下的紫外‐可见(UV‐Vis)吸收特性,其光谱如图4所示。在溶液中,TTIC和TTIC‐M在550至800纳米范围内表现出强烈的吸收,吸收峰分别位于750和739纳米,最大摩尔消光系数分别为1.48 × 10⁵ M⁻¹ cm⁻¹和1.42 × 10⁵ M⁻¹ cm⁻¹。从溶液到薄膜,吸收光谱变得更宽,TTIC和TTIC‐M的最大吸收峰分别红移61和37纳米,表明在固态中存在强烈的π‐π分子间相互作用。TTIC和TTIC‐M薄膜的吸收起始点分别位于885和862纳米,根据公式Eg,opt=1240/λonset计算,对应的光学带隙分别为1.40和1.44电子伏特。
TTIC和TTIC‐M均与PBDB‐T表现出较强的互补吸收。宽广的光吸收带和高吸收系数有助于捕获更多的太阳光子,并在器件中产生高的Jsc。

3.3. 电化学性质
TTIC、TTIC‐M和PBDB‐T的电化学性质通过在含0.1 M Bu₄NPF₆的CH₃CN溶液中,使用碳工作电极进行循环伏安法(CV)平行测定。以二茂铁/二茂铁阳离子(Fc/Fc⁺)氧化还原电对作为内参比标准,其电位为0.10 V。假设Fc/Fc⁺在真空中的绝对能级为4.8 eV。电化学能级根据公式E = ‐e[Eox/red + 4.80 ‐ E(Fc/Fc⁺)]计算。如图5(a)所示,TTIC、TTIC‐M和PBDB‐T的起始氧化电位(Eox)分别为1.05、0.98和0.66 V,对应的HOMO能级分别为‐5.75、‐5.68和‐5.36 eV。
TTIC和TTIC‐M的起始还原电位(Ered)分别为‐0.78和‐0.84 V。TTIC和TTIC‐M的电化学LUMO能级分别计算为‐3.92 eV和‐3.86 eV。光学LUMO能级通过公式ELUMO = EHOMO + Eg,opt计算,TTIC、TTIC‐M和PBDB‐T的值分别为‐4.35、‐4.24和‐3.55 eV。所有这些数据汇总于表1中,TTIC、TTIC‐M和PBDB‐T的光学能级如图4 (b)所示。值得注意的是,由于甲基的弱给电子特性,甲基取代的TTIC‐M的HOMO和最低未占分子轨道能级高于TTIC。因此,基于TTIC‐M的器件的Voc应高于基于TTIC的器件。

| 化合物 | λmax薄膜 (nm) | λedge薄膜 (nm) | Eg,opt (eV) | HOMO (eV) | 最低未占分子轨道 a (eV) | 最低未占分子轨道 b (eV) |
|---|---|---|---|---|---|---|
| TTIC | 811 | 885 | 1.40 | -5.75 | -4.35 | -3.92 |
| TTIC‐M | 776 | 862 | 1.44 | -5.68 | -4.24 | -3.86 |
| PBDB‐T | 633 | 685 | 1.81 | -5.36 | -3.55 | ‐2.92[24] |
a 光学LUMO能级
b 电化学LUMO能级
3.4. 光伏性能
器件结构为 ITO(氧化铟锡)/ZnO(氧化锌)(30 纳米)/活性层(85 纳米)/ MoO₃ (8.5 纳米)/Ag(100 纳米)的体异质结光伏器件被用于研究以 TTIC 和 TTIC‐M 作为受体、PBDB‐T 作为供体时的光伏性能。系统地研究了一系列条件,包括供体与受体质量比、活性层厚度、添加剂和热退火(表 S1‐S5)。
表2总结了基于PBDB‐T:TTIC(1:1.5,w/w)和PBDB‐T:TTIC‐M(1:1.5, w/w)器件的相应光伏参数。在AM 1.5G(100 mW cm⁻²)光照条件下,具有最佳光伏性能的器件的电流密度‐电压(J‐V)曲线如图6所示。优化后,基于 PBDB‐T:TTIC的器件实现了9.73%的光电转换效率(PCE),开路电压(Voc) 为0.77 V,填充因子(FF)为0.62,短路电流密度(Jsc )达到显著的20.39 mA cm⁻²。加入0.15%(体积比)DIO并对共混薄膜在 125 °C退火5分钟之后, Jsc和PCE进一步提升至22.26 mA cm⁻²和10.61%。如预期所示,基于 PBDB‐T:TTIC‐M的器件表现出更高的Voc (0.81 V),光电转换效率达到 9.62%,其Jsc为18.19 mA cm⁻² ,填充因子为0.65。加入0.2%(体积比)DIO并对共混薄膜在175 °C退火5分钟后,Jsc和FF显著提高至20.51 mA cm⁻²和0.70。得益于更高的Voc、优异的Jsc以及高填充因子,基于 PBDB‐T:TTIC‐M的器件实现了进一步提升的11.48%光电转换效率。据我们所知,11.48%是目前基于窄带隙(<1.45 eV)非富勒烯受体器件的最高光电转换效率。
| 器件 | DIO退火 | 伏特 oc (伏特) | J sc (毫安/平方厘米) | FF | 光电转换效率 最佳/平均 a (百分比) |
|---|---|---|---|---|---|
| TTIC | – | 0.77 | 20.39 | 0.62 | 9.73/9.57 |
| TTIC 优化后 | 0.15% 125 °C, 5分钟 | 0.77 | 22.26 | 0.62 | 10.61/10.34 |
| TTIC‐M | – | 0.81 | 18.19 | 0.65 | 9.62/9.60 |
| TTIC‐M 优化后 | 0.20% 175 °C,5分钟 | 0.80 | 20.51 | 0.70 | 11.48/11.40 |
a 平均PCE根据5个器件计算得出。
基于PBDB‐T:TTIC和PBDB‐T:TTIC‐M的器件的外量子效率(EQE)曲线如图 6b所示。所有器件均表现出较强的在300到900纳米范围内的光电响应。通过对外量子效率曲线积分推导出的Jsc值 与电流‐电压测量结果吻合良好(误差在5%以内)。基于TTIC的优化器件表现出略高于TTIC‐M的外量子效率,这与其较高的Jsc一致。
 外量子效率 ,(c)优化器件的短路电流密度(Jsc)与光强的关系)
外量子效率 ,(c)优化器件的短路电流密度(Jsc)与光强的关系)
3.5. 双分子复合动力学
我们还通过测量光电流密度(Jsc)随光强(P)的变化来研究电荷复合行为。J sc与P的关系为Jsc= k PS。若所有自由载流子在复合前均被电极完全收集,则S应等于1。根据图6(c)计算得到基于TTIC和TTIC‐M的优化器件的S值分别为0.94 和0.98,表明基于TTIC‐M的器件中双分子复合被更有效地抑制,这可能是TTIC‐M基器件具有更高填充因子的原因之一。[28, 29]
3.6. 传输特性
为了进一步理解基于TTIC和TTIC‐M的聚合物太阳能电池之间不同的光伏性能, 采用ITO/PEDOT:PSS/活性层/Au结构的仅空穴器件和仅电子器件ITO/ZnO/活性层/Al 器件被制备。空穴迁移率 (μh) 和电子迁移率 (μe) 通过空间电荷限制电流 (SCLC) 方法进行评估。如图 S3 所示,优化后基于TTIC的器件的空穴迁移率和电子迁移率分别计算为 9.24 × 10⁻⁵ 和 1.07 × 10⁻⁴ cm² V⁻¹ s⁻¹。基于TTIC‐M的器件表现出的空穴迁移率和电子迁移率分别为 1.25 × 10⁻⁴ 和 1.33 × 10⁻⁴ cm² V⁻¹ s⁻¹。相对于基于TTIC的器件,TTIC‐M 显示出更平衡的电子/空穴迁移率 (μe/μh=1.06),这有利于获得较高的填充因子 FF。空穴和电子迁移率数据见表 3。
| 器件 | μh(cm² V⁻¹ s⁻¹) | μe(cm² V⁻¹ s⁻¹) | μe /μh | 厚度 (纳米) |
|---|---|---|---|---|
| TTIC 优化后 | 9.24 × 10⁻⁵ | 1.07 × 10⁻⁴ | 1.15 | 85 |
| TTIC‐M 优化后 | 1.25 × 10⁻⁴ | 1.33 × 10⁻⁴ | 1.06 | 86 |
3.7. 薄膜形貌
原子力显微镜(AFM)和透射电子显微镜(TEM)被用于研究活性层的薄膜形貌。PBDB‐T:TTIC和PBDB‐T:TTIC‐M的原始及优化后的共混薄膜如图7所示。与原始共混薄膜相比,加入DIO并进行热退火后,表面形貌变得更加均匀,相区尺寸显著减小。基于PBDB‐T:TTIC和PBDB‐T:TTIC‐M的共混薄膜在优化后表现出均匀但略微粗糙的表面,这可能源于薄膜退火后结晶性的提高。PBDB‐T:TTIC和PBDB‐T:TTIC‐M共混薄膜的均方根(RMS)粗糙度值分别为 5.28 nm和2.52 nm。图8显示了PBDB‐T:TTIC和PBDB‐T:TTIC‐M共混薄膜。对于TTIC和TTIC‐M,均可以在原始和优化后的共混薄膜中清晰观察到分散良好的纤维结构。这些纳米纤维是在薄膜干燥过程中由PBDB‐T[30]聚集形成的。经过优化后,PBDB‐T:TTIC共混薄膜中纳米纤维的宽度从18至38纳米增加,而PBDB‐T:TTIC‐M共混薄膜中的纳米纤维宽度从16至26纳米增加。适当的纳米纤维宽度有利于载流子的生成和传输。
 的 (a) PBDB‐T:TTIC 铸态, (b) PBDB‐T:TTIC 优化后, (c) PBDB‐T:TTIC‐M 铸态 和 (d) PBDB‐T:TTIC‐M 优化后)
的 (a) PBDB‐T:TTIC 铸态, (b) PBDB‐T:TTIC 优化后, (c) PBDB‐T:TTIC‐M 铸态 和 (d) PBDB‐T:TTIC‐M 优化后)
 PBDB‐T:TTIC 优化后、(c) PBDB‐T:TTIC‐M 铸态和 (d) PBDB‐T:TTIC‐M 优化后)
PBDB‐T:TTIC 优化后、(c) PBDB‐T:TTIC‐M 铸态和 (d) PBDB‐T:TTIC‐M 优化后)
4. 结论
两种新型非富勒烯受体(TTIC和TTIC‐M)被设计并合成,它们以稠合六环单元DTCTT作为富电子单元,1,1‐二氰亚甲基‐3‐茚酮作为缺电子单元。TTIC和TTIC‐M的吸收边延伸至近红外区域(900纳米),其光学带隙分别低至1.40和1.44电子伏特。当与宽带隙聚合物给体PBDB‐T共混时,基于TTIC的器件的光电转换效率达到10.61%,同时表现出优异的 Jsc(22.26 毫安/平方厘米)。由于甲基取代能够提升受体的LUMO能级,正如预期,基于TTIC‐M的器件表现出更高的Voc。基于TTIC‐M的器件实现了11.48%的高光电转换效率,其Jsc为20.51 毫安/平方厘米,Voc为0.80 伏特,填充因子为0.70。
基于PBDB‐T:TTIC‐M的器件。据我们所知,11.48%是迄今为止基于窄带隙 (<1.45 eV)非富勒烯受体的此类器件的最高性能,表明TTIC和TTIC‐M是未来实际应用中有前景的受体材料。

















 3346
3346

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









