







 本文介绍了两种方法解决DNA排序问题,法1是手写版,通过ASCII值比较基因的无序度;法2则是教师版本,利用嵌套循环和ASCII值计数。文章详细解释了奇技淫巧,包括如何计算无序度、存储基因和索引操作。
本文介绍了两种方法解决DNA排序问题,法1是手写版,通过ASCII值比较基因的无序度;法2则是教师版本,利用嵌套循环和ASCII值计数。文章详细解释了奇技淫巧,包括如何计算无序度、存储基因和索引操作。
题目

Code
-法1(自己肝出来的)
n,m=map(int,input().split())
gene_lib=[]
inver=[]
for i in range(m):
gene=input()
gene_lib.append(gene)
count=0
for j in range(n):
asc=ord(gene[i])
for h in range(j,n):
if asc>ord(gene[h]):
count+=1
inver.append(count)
inver_index=sorted(range(m),key=lambda i:inver[i])
for i in inver_index:
print("".join(gene_lib[i]))
代码解析
手写版

法2(teacher版)
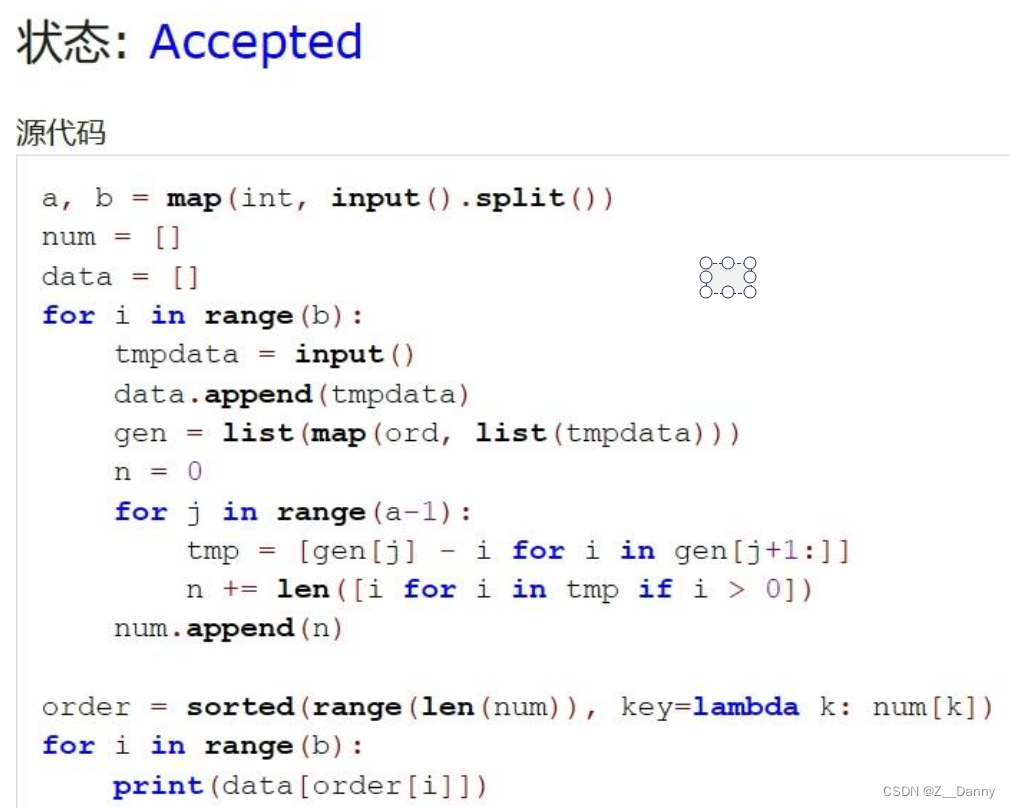
代码解析
其实思路和法1差不多,就是表达方式不太一样吧
解题思路
主要用到A-Z的ascii值从小到大。从上到下第一个循环,作为每行输入。第二个循环是用于将gene的每个ASCII值,第三个循环是用于与它右边的字母的ASCII值进行对比,如果发现一次,计数器+1。然后用一个列表把每行gene的无序度记录下来,再对这个列表进行排序返回索引值,把这个索引值用到原来存放所有gene的进行输出。
奇技淫巧
-
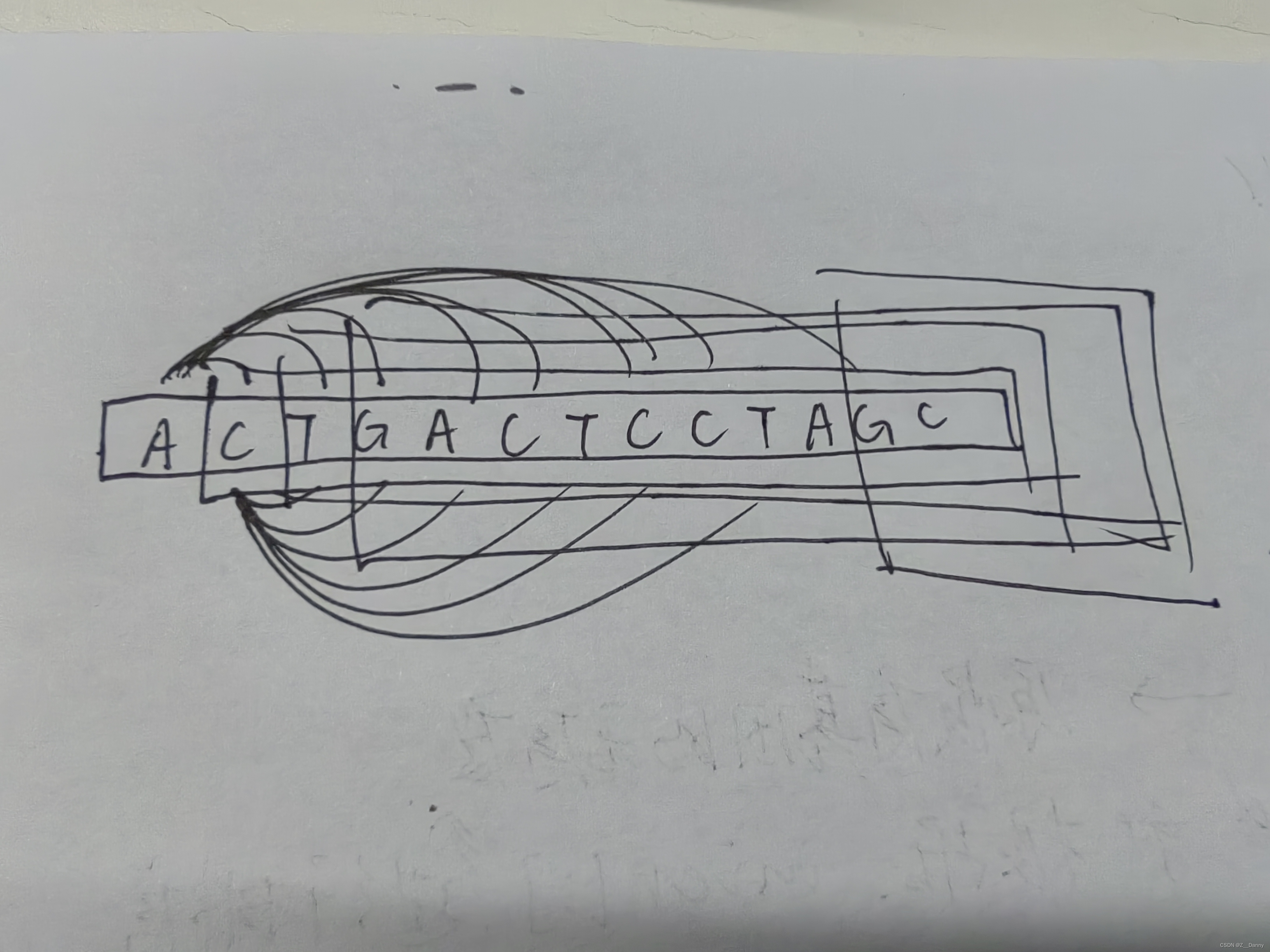
Q:怎么计算一条基因的无序度呢?以什么为标准嘞?
–>因为这个是通过26字母的先后判断大小,我们就可以想到用ASCII值进行比较,刚好排在字母表后的字母的ASCII值会比前面的大 -
Q:既要又要怎么办?既要比较又要计数怎么安排?
–>比较的时候需要遍历,每一个基因,但是可以只比较它后面的和它就可以了,只要后面的比它小就计数+1,有种倒着来的感觉。所以就是循环每一个基因,然后又嵌套一个从它本身往后的一个遍历进行比较。
-
Q:需要储存哪些结果呢?
–>需要首先存储每一条输入的基因,进入基因库列表,因为输出要求重新提取这些基因序列;
其次还需要存储每条基因的无序度,便于后续对它们进行无序度的排序 -
Q:怎么把获得的结果可以和输出联系起来?
–>因为我们的无序度是按照原基因顺序对应着存储的,所以我们使用sorted的函数时,可以以无序度的值比较,用len(m)进行排序绑定各个无序度进行排序,实现索引的排序,最后,按照索引在基因库找基因序列输出即可


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









