




蛋白质之间的相互作用网络是细胞生命活动的基础,许多疾病的发生往往与这些相互作用的异常有关。传统的蛋白质互作研究方法存在一定局限:它们通常只能捕获高亲和力的静态相互作用,且需要在非生理条件下进行体外实验。为了克服这些不足,科学家们开发了邻近标记技术,可以在活细胞内动态标记目标蛋白周围的邻近蛋白,从而揭示更完整的蛋白质互作网络。其中,TurboID是近年发展起来的一种高效邻近标记酶,已在哺乳动物、植物、微生物等多种体系中得到应用。同时,定量蛋白质组学技术的进步使得我们能够对不同条件下的蛋白质进行精确的相对或绝对定量分析。将TurboID邻近标记与定量蛋白质组学相结合,为绘制高分辨率的蛋白质相互作用网络提供了强大的新工具。
一、TurboID邻近标记技术的原理
TurboID是一种经过定向进化改造的生物素连接酶,最初源于大肠杆菌的BirA蛋白。与早期的BioID酶相比,TurboID的催化活性显著提高,能够在更短时间内标记目标蛋白周围的邻近分子。其工作原理是:将TurboID酶与目标蛋白融合表达,在细胞内加入生物素(biotin)后,TurboID利用ATP将生物素转化为高反应性的生物素-5’-AMP中间体,并在酶的作用半径内将生物素共价连接到邻近的蛋白上。被标记的蛋白质随后可通过链霉亲和素磁珠富集,并进行质谱鉴定。由于生物素标记在活细胞内完成,这一过程能够捕获瞬时的、弱亲和力的相互作用,以及发生在特定亚细胞定位的互作,从而弥补了传统方法的不足。
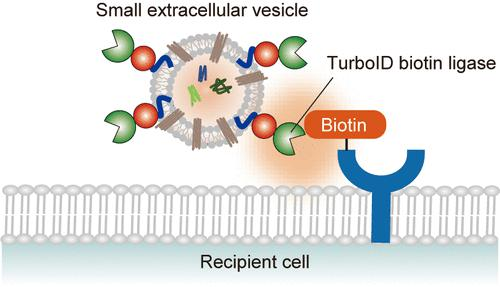
TurboID技术原理图
二、定量蛋白质组学的原理
定量蛋白质组学旨在对不同样品中的蛋白质进行相对或绝对的定量比较,以研究蛋白质表达水平的变化和相互作用的强度差异。目前常用的定量策略包括标记定量和非标记定量两大类。
- 标记定量法 :在样品处理过程中引入稳定同位素标记,使不同样品中的同一蛋白质带有可区分的“重”或“轻”同位素标签。常用的方法有: 代谢标记 (如SILAC,在细胞培养基中加入含稳定同位素的氨基酸)、 化学标记 (如iTRAQ、TMT,通过化学试剂对肽段进行同位素标记)等。标记后的样品可混合进行质谱分析,通过检测同位素峰的强度比来计算蛋白质的相对丰度。标记法的优点是定量准确、重复性好,适合多组样品的平行比较。
- 非标记定量法 :不使用同位素标签,而是通过质谱数据本身的信息进行定量。例如 光谱计数 (统计每个蛋白质被鉴定的肽段质谱信号次数)或 峰面积积分 (测量肽段色谱峰的面积)等方法。非标记定量操作相对简单,不受样品来源限制,但对质谱仪器的稳定性和数据处理算法要求较高。
无论采用何种方法,定量蛋白质组学通常结合高分辨液相色谱-串联质谱(LC-MS/MS)技术对肽段进行分离和鉴定,并利用生物信息学软件对质谱数据进行分析,以计算蛋白质的相对丰度差异。定量蛋白质组学已广泛应用于比较不同生理或病理状态下的蛋白质表达谱、筛选生物标志物,以及解析药物处理后的蛋白变化等研究中。

高分辨率液相色谱-串联质谱(LC-MS/MS)系统实验室仪器
三、TurboID与定量蛋白质组学联用的技术路线
在邻近标记研究中,定量蛋白质组学尤为重要。由于邻近标记会标记目标蛋白附近的众多蛋白,包括一些非特异性结合的背景蛋白,通过引入定量分析可以区分特异性互作蛋白和背景噪音。通常的做法是在实验中设置对照(例如仅表达TurboID标签而不连接目标蛋白的细胞),通过比较实验组与对照组中各蛋白质的定量信号强度,筛选出在实验组中显著富集的蛋白作为候选互作蛋白。这种定量比较大大提高了邻近标记结果的可靠性,使研究者能够更准确地绘制出目标蛋白的互作网络。
将TurboID邻近标记与定量蛋白质组学相结合,可以在活细胞内高效地捕获并定量目标蛋白的邻近互作蛋白。其典型技术路线包括以下步骤:
1.构建融合表达载体:将TurboID编码序列与目标蛋白基因融合,构建表达质粒。例如,在KRAS研究中,可将TurboID连接到KRAS蛋白的N端或C端,并引入HA等标签以便检测。构建完成后,通过测序验证序列正确性。
2.建立稳定表达细胞系:将上述融合表达载体导入目标细胞(如哺乳动物细胞系),通过抗生素筛选获得稳定表达TurboID-目标蛋白融合体的细胞克隆。同时设置对照组,如仅表达TurboID标签或无关蛋白融合的细胞,用于后续定量比较。
3.邻近标记反应:在稳定细胞系中加入生物素底物,孵育一定时间,让TurboID催化生物素标记周围蛋白。孵育时间根据目标蛋白性质优化,通常在几分钟到1~2小时范围内。例如,有研究对HEK293T细胞表达的KRAS-TurboID融合蛋白加入50 μM生物素孵育2小时,实现了对KRAS邻近蛋白的有效标记。
4.裂解与富集:标记完成后,裂解细胞提取总蛋白,然后利用链霉亲和素偶联的磁珠对生物素化的蛋白进行亲和富集。经过充分洗涤去除未结合的杂质后,将富集到的蛋白样品进行后续分析。
5.定量蛋白质组学分析:将富集的蛋白样品进行酶解消化成肽段,然后通过LC-MS/MS进行分离和鉴定。为了定量比较,可采用标记定量策略(如将实验组与对照组样品用iTRAQ/TMT标记后混合进样)或非标记定量策略(如对实验组和对照组分别进行质谱分析,通过软件对齐色谱峰定量)。质谱数据经数据库检索鉴定蛋白质,并计算各蛋白在实验组相对于对照组的富集倍数或丰度比值。
6.数据处理与互作网络构建:利用生物信息学工具对定量结果进行分析,筛选出显著富集的蛋白质作为目标蛋白的邻近互作候选。通常以富集倍数和统计学显著性(如p值)作为标准,例如FC≥2且p <0.05的蛋白被认为是特异性互作蛋白。随后,可进一步对这些候选蛋白进行功能富集分析(如GO、KEGG通路富集),并构建蛋白质互作网络图谱,以揭示目标蛋白参与的生物学过程和信号通路。
7.验证与功能研究:对筛选出的关键互作蛋白,可通过免疫共沉淀、免疫荧光共定位等传统方法进行验证。此外,结合基因敲除、过表达或小分子干预等手段,研究这些互作蛋白在目标蛋白功能中的作用,从而阐明其生物学意义。
通过以上流程,TurboID邻近标记与定量蛋白质组学的联用能够在活细胞环境中高效、系统地绘制目标蛋白的相互作用网络,并量化不同条件下互作的变化。这一技术路线已被成功应用于多种研究,包括细胞器蛋白组的鉴定、信号复合物的解析,以及疾病相关蛋白互作网络的绘制等。
四、TurboID与定量蛋白质组学在KRAS研究中的应用实例
KRAS基因是RAS原癌基因家族中最常见的成员,其突变在多种人类癌症中高频发生,是癌症发生的重要驱动因素之一。正常情况下,KRAS蛋白作为分子开关,在结合GTP时激活下游信号,结合GDP时失活。大量研究表明,不同KRAS突变体可能通过影响其与上下游蛋白的相互作用,从而改变下游信号通路的激活模式和强度。因此,绘制不同KRAS突变体的相互作用网络并比较其异同,将有助于揭示突变特异性的致癌机制,并为精准医疗策略提供依据。

本研究通过构建稳定表达HA-TurboID标记的KRAS野生型和G12C/D/V突变体的HEK293T 细胞系,运用TurboID邻近标记和定量蛋白质组学技术,系统绘制了KRAS及其突变体的蛋白质相互作用网络,发现突变体与野生型在多条关键代谢通路中的相互作用蛋白存在显著差异,且均表现出与LZTR1结合减少、与LAMTOR1招募增强的共有变化,揭示了KRAS G12C、G12D和G12V突变通过改变蛋白质相互作用网络影响细胞代谢状态,进而促进肿瘤增殖和免疫逃逸的机制,为精准治疗提供了LZTR1和LAMTOR1这两个新的潜在靶点。TurboID邻近标记结合定量蛋白质组学的方法在KRAS研究中取得了丰硕成果。它不仅高效地绘制了KRAS及其突变体的相互作用网络,还从中解析出驱动癌症发生的关键机制和潜在治疗靶点。这些发现凸显了该联用技术在癌症研究中的强大威力,也为其他疾病相关蛋白互作网络的研究提供了范例。
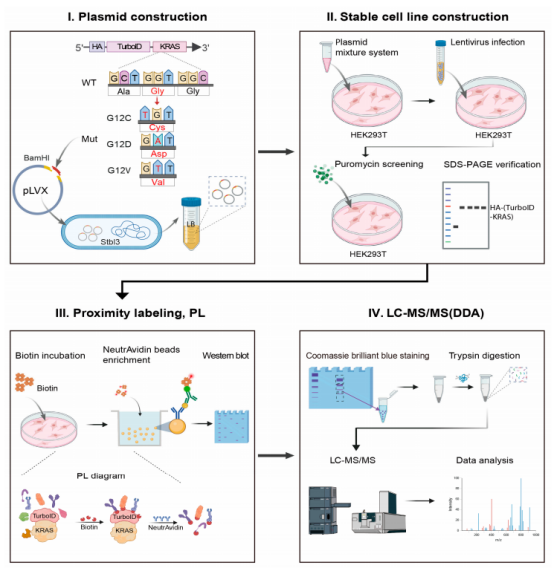
研究设计图
五、总结
TurboID邻近标记技术与定量蛋白质组学的结合,代表了当前蛋白质相互作用研究领域的前沿方向。通过在活细胞内实时捕获邻近蛋白并进行精确的定量比较,研究者能够以前所未有的深度和广度揭示蛋白质互作网络的动态变化。本文围绕KRAS及其突变体的研究案例表明,该联用方法不仅可以鉴定大量新的互作蛋白,还能结合生物信息学分析阐明这些互作所涉及的分子机制和生物学功能。在KRAS研究中,我们看到不同突变如何通过改变互作网络来重编程代谢和影响肿瘤微环境,从而推动癌症的发生发展。这些成果不仅加深了对KRAS致癌机制的认识,也为开发更有效的靶向治疗策略提供了新思路。

















 1541
1541

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









