







本文较长,建议先收藏后随时查看!以后我们将更新更多计算解析,欢迎关注!
01
背景
传统电池的石墨负极和金属氧化物正极在高充电速率下容易出现性能退化,限制了快速充电的实现。为解决这些问题,研究者们开始探索新型二维(2D)材料,这些材料因其高比表面积、优异的电子性能和潜在的快速充电能力而备受关注。
本研究,发表在《Physical Chemistry Chemical Physics》(2023年)上的文章《Toward efficient electrodes for a high-performance fast-charge Li-ion battery: molecular dynamics simulation and DFT calculations》(DOI:10.1039/d2cp06020e),聚焦于一种新型锂离子纳米电池设计,采用铜苯六硫醇(Cu-BHT)金属有机框架(MOF)作为正极,磷烷(黑磷单层)作为负极。

Cu-BHT是一种二维金属有机框架,具有高导电性和稳定性,是理想的锂存储材料。磷烷因其高比表面积和锂嵌入后导电性的增强,被认为是潜在的负极材料。本研究通过分子动力学(MD)模拟和密度泛函理论(DFT)计算,探索了该电池在不同外加电场下的充电动态,以揭示锂离子迁移机制和电极材料的电子结构特性。
02
方法
本研究采用两种主要的计算方法:分子动力学(MD)模拟和密度泛函理论(DFT)计算,分别用于研究锂离子在电池系统中的动态行为和电极材料的电子结构。
分子动力学模拟(MD)
软件与设置:使用LAMMPS软件包进行模拟,初始配置通过PACKMOL生成,模拟框尺寸为200 × 58 × 38 ų,采用周期性边界条件。
电极材料:负极为由5632个磷原子组成的7层磷烷晶体,正极为由588个铜原子、1099个硫原子和924个碳原子组成的7层Cu-BHT MOF。
电解质:使用1.5 M LiPF₆溶于乙烯碳酸酯(EC),并加入铜、铝和SiO₂作为集流体以维持结构完整性。
模拟过程:系统首先在NVT系综下进行热平衡,从5 K逐渐升至293 K。随后在NPT系综下施加0.25 V Å⁻¹和1.5 V Å⁻¹的外加电场,模拟充电过程。
数据分析:通过计算径向分布函数(RDF)、均方位移(MSD)和扩散系数,分析锂离子的迁移行为和系统内的相互作用。
密度泛函理论计算(DFT)
计算工具:使用Vienna ab initio simulation package(VASP)软件,采用广义梯度近似(GGA)和Perdew-Burke-Ernzerhof(PBE)泛函,结合投影增强波(PAW)方法,平面波截止能量为400 eV。
系统设置:Cu-BHT使用1 × 1 × 1超胞,磷烷使用3 × 3 × 1超胞,计算中考虑了范德华力校正(DFT-D3)。
研究内容:分析锂在Cu-BHT和磷烷上的吸附位点、吸附能量,以及锂嵌入对材料电子结构(如能带结构和态密度)的影响。
添加下方微信好友,立即咨询计算


电话/微信:13564914850
03
模拟
充电模拟
研究者在MD模拟中施加了两种不同强度的外加电场(0.25 V Å⁻¹和1.5 V Å⁻¹),以观察锂离子从Cu-BHT正极到磷烷负极的迁移行为。在较低电场(0.25 V Å⁻¹)下,13个锂离子成功到达负极;而在较高电场(1.5 V Å⁻¹)下,17个锂离子迁移至负极,表明较高电场显著加速了充电过程。
这些结果通过图表展示,例如Figure 7展示了不同电场下到达负极的锂原子数量,清晰反映了电场强度对充电速率的影响。然而,模拟结果也显示,较高电场可能导致系统稳定性下降,表现为系统能量的波动,如Figure 4所示,图中展示了不同电场下系统能量的随时间演化。
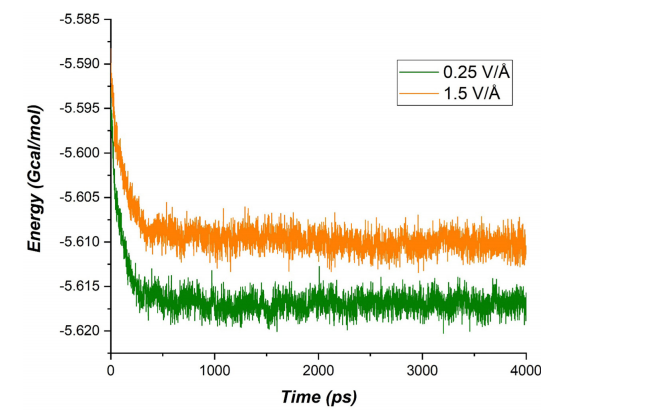
图4 不同电场强度对能量随时间演变的影响。
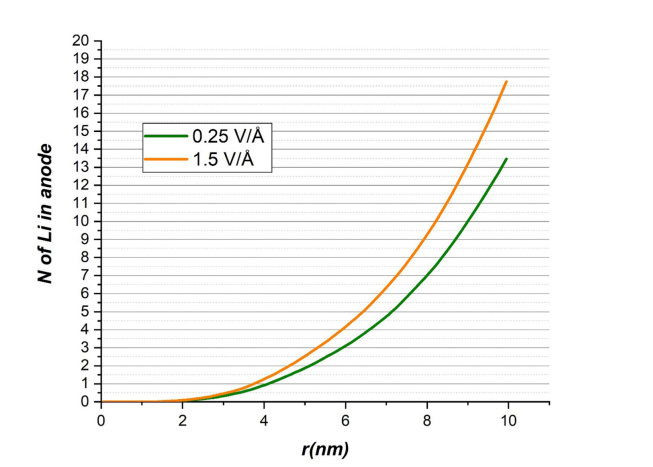
图7 不同电场强度对到达阳极的锂原子数量的影响。
离子分布与迁移
通过径向分布函数(RDF)分析,研究者观察到锂离子与磷烷之间的相互作用峰值,分别位于1.4 nm和3.8 nm,确认了锂离子的迁移路径。这些结果通过Figure 5展示,RDF图清晰呈现了不同电场下锂离子与磷烷的分布和动态行为。
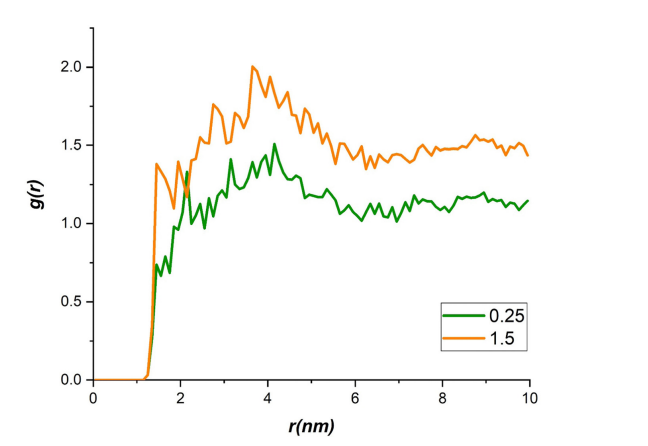
图5 在不同电场(0.25和1.5 V Å⁻¹)下,锂离子相对于磷烷的径向分布函数。
均方位移(MSD)和扩散系数分析进一步表明,较高电场下锂离子迁移速度加快,但由于离子间相互作用增强,整体移动性略有下降。这些动态行为通过Figure 9展示,图中呈现了锂离子的MSD曲线及相应的扩散系数,反映了电场对离子移动性的影响。
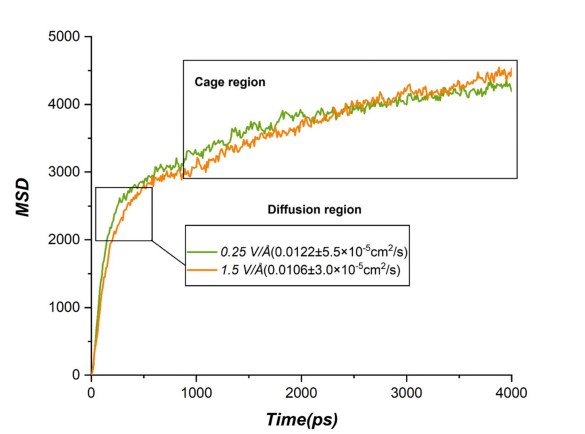
图9 在施加电场下的研究系统中,锂离子的均方位移(MSD)随模拟时间变化及其对应的扩散系数(D值)。
此外,Figure 3提供了充电过程中电池的快照,展示了锂离子从正极向负极迁移的过程,例如在18 ps时无锂离子到达负极,而在4000 ps时锂离子已显著嵌入负极。
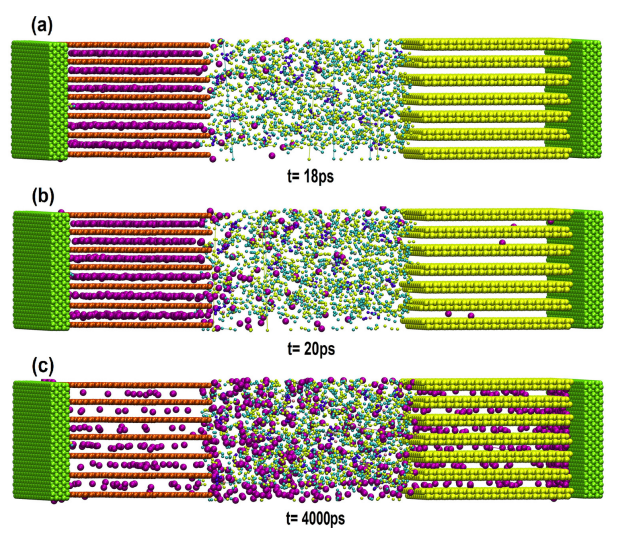
图3 纳米电池首次充电过程中拍摄的快照。(a) 18 ps时的锂离子电池,阳极中未存储锂原子。(b) 20 ps时的锂离子电池,磷烷层将成为锂原子的宿主。(c) 4000 ps时电池充电的快照。
电子结构分析
DFT计算提供了电极材料的电子结构信息。锂在Cu-BHT上的吸附能量为-3.21 eV(最稳定位点为六元环,C位点,如Figure 11和Figure 12所示),在磷烷上的吸附能量为-2.06 eV(最稳定位点为空位,H位点,如Figure 14和Figure 15所示)。
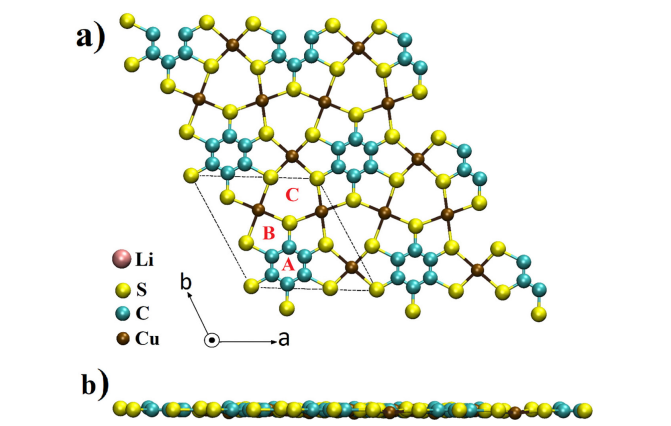
图11 2D Cu-BHT MOF结构的(a) 顶视图和(b) 侧视图。字母表示Li⁺吸附的不同位置,虚线为单位细胞。
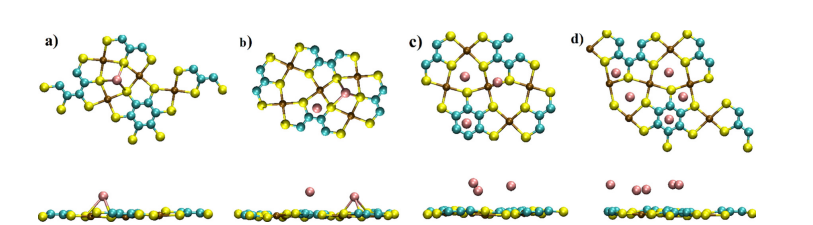
图12 完全优化的结构的顶视图和侧视图:(a) C配置,(b) BC配置,(c) ABC配置,(d) Li5-MOF配置。
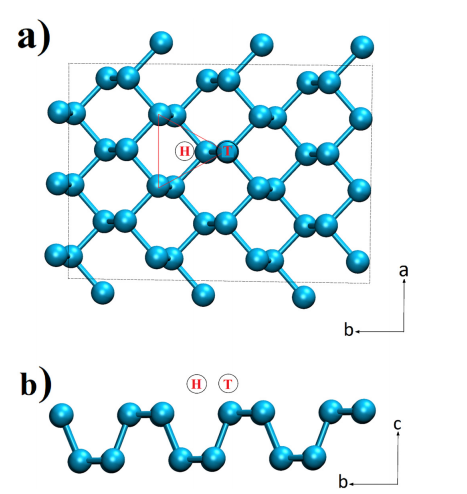
图14 磷烷单层的(a) 顶视图和(b) 侧视图。标记为H(空心)和T(顶部)的虚线圆圈表示锂原子吸附的两个代表性位点。虚线为单位细胞。
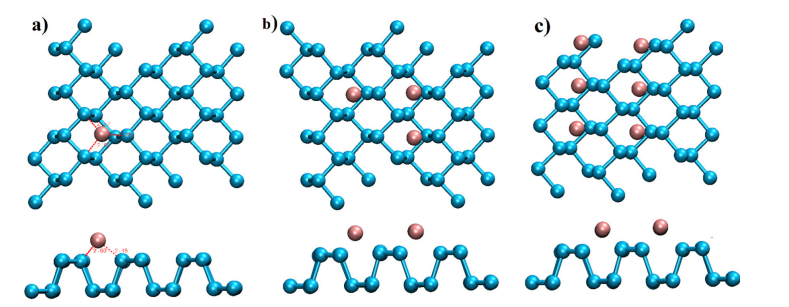
图15 完全优化的结构的顶视图和侧视图:(a) H配置,(b) Li3-磷烷配置,(c) Li6-磷烷配置。
Cu-BHT表现出金属导电性,锂吸附后导电性进一步增强,如Figure 13所示,图中展示了Cu-BHT在不同锂吸附配置下的能带结构和态密度(DOS),表明其金属特性。
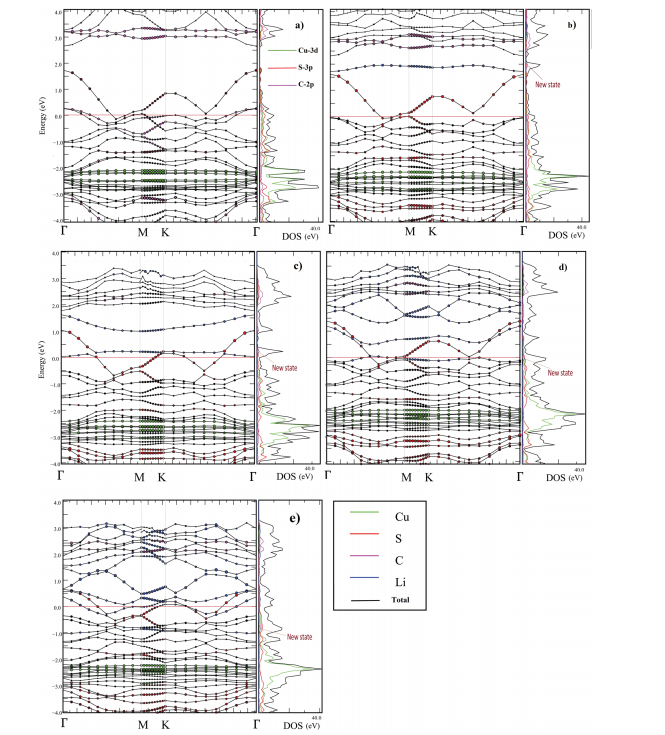
图13 (a) 新鲜Cu-BHT,(b) C配置,(c) BC配置,(d) ABC配置,(e) Li5-Cu-BHT配置的能带结构和部分态密度(DOS)。费米水平由红色竖线标记并设为零。
磷烷的带隙为0.9 eV,锂嵌入后带隙减小,导电性提升,如Figure 16所示,图中展示了磷烷在不同锂吸附状态下的能带结构和DOS。这些结果表明两种材料在快速充电中的优异性能。
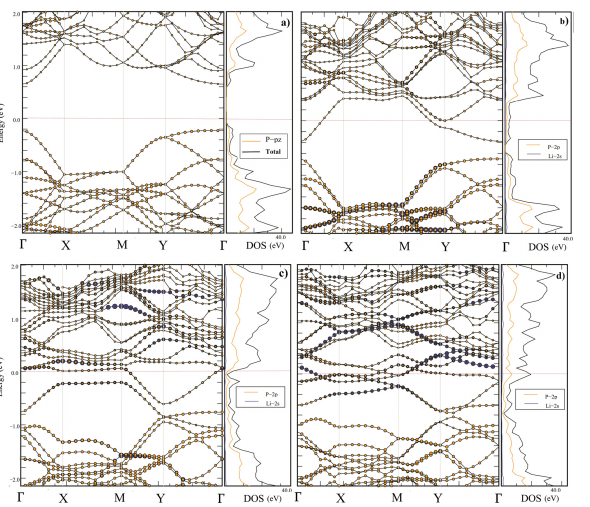
图16 (a) 新鲜磷烷,(b) H配置,(c) Li3-磷烷配置,(d) Li6-磷烷配置的能带结构和部分态密度(DOS)。
04
结论
论文贡献点
充电动态研究:通过MD模拟,系统分析了Cu-BHT和磷烷电极纳米电池在不同电场下的锂离子迁移行为,揭示了快速充电的潜力。
材料性能分析:DFT计算确认Cu-BHT和磷烷具有优异的锂吸附能力和导电性,适合作为高性能电极材料。
方法论创新:结合MD和DFT方法,提供了对电池性能的全面理解,为未来电池设计提供了理论参考。
局限性
模拟时间限制:MD模拟的时间尺度可能不足以捕捉某些长时间尺度的动态过程。
模型简化:模拟系统可能未完全反映真实电池环境的复杂性,如电解质的复杂化学反应。
样本数量:研究仅涉及特定电极材料组合,结果的普适性需进一步验证。
免责声明


















 29
29

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











