




 UCell是一个工具,最初设计用于通过细胞类型签名打分进行细胞注释,也可以用于其他基因集的打分。它在Seurat对象的元数据列中存储每个细胞的得分。提供的示例包括简单快速的使用方法和完整的Seurat标准流程结合UCell进行细胞类型和亚群鉴定的流程。
UCell是一个工具,最初设计用于通过细胞类型签名打分进行细胞注释,也可以用于其他基因集的打分。它在Seurat对象的元数据列中存储每个细胞的得分。提供的示例包括简单快速的使用方法和完整的Seurat标准流程结合UCell进行细胞类型和亚群鉴定的流程。
ucell的设计本意是通过cell type signature打分进行细胞注释,但一样可以用于其它gene set的打分
首先做好gene signature list,与seurat流程相整合。对每个细胞计算每个条目的得分,每个条目的得分会存储在seurat object- metadata里的列。
demo1: 简单快速用法
注意:ucell直接在count上走流程,可视化的时候才normalize scale等.但其他的demo都是先走标准流程。
library(remotes)
remotes::install_github("carmonalab/UCell")
library(UCell)
set.seed(123)
###加载示例数据集
library(GEOquery)
cached.object <- "demodata.rds"
if (!file.exists(cached.object)) {
geo_acc <- "GSE115978"
options(timeout = max(1000, getOption("timeout")))
gse <- getGEO(geo_acc)
getGEOSuppFiles(geo_acc)
exp.mat <- read.csv(sprintf("%s/GSE115978_counts.csv.gz", geo_acc), header = T,
row.names = 1, sep = ",")
saveRDS(exp.mat, cached.object)
} else {
exp.mat <- readRDS(cached.object)
}
###define gene signatures
signatures <- list(Immune = c("PTPRC"),
Macrophage = c("CTSB", "C1QB", "LAPTM5", "TYROBP", "PSAP", "C1QA", "HLA-DRA", "CTSD", "NPC2", "FCER1G"),
Tcell = c("CD3D", "CD3E", "CD3G", "CD2"),
Bcell = c("MS4A1", "CD79A", "CD79B", "CD19", "BANK1"),
Myeloid_cell = c("CD14", "LYZ", "CSF1R", "FCER1G", "SPI1", "LCK-"),
Stromal = c("MMP2", "COL1A1", "COL1A2", "COL3A1", "LUM", "DCN"))
###perform ucell
u.scores <- ScoreSignatures_UCell(exp.mat, features = signatures)
u.scores[1:8, 1:2]
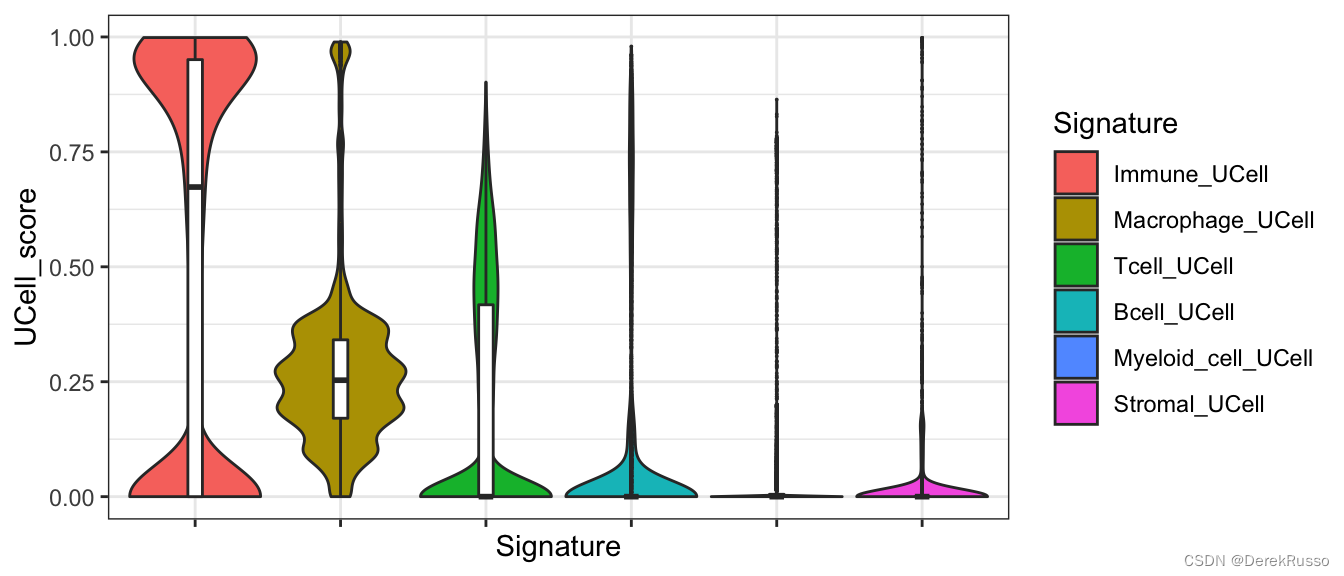
###show the distrobution of the scores
library(reshape2)
library(ggplot2)
melted <- reshape2::melt(u.scores)
colnames(melted) <- c("Cell", "Signature", "UCell_score")
p <- ggplot(melted, aes(x = Signature, y = UCell_score)) + geom_violin(aes(fill = Signature),
scale = "width") + geom_boxplot(width = 0.1, outlier.size = 0) + theme_bw() +
theme(axis.text.x = element_blank())
p
###ucell seurat
library(Seurat)
seurat.object <- CreateSeuratObject(counts = exp.mat, project = "JerbyArnon")
seurat.object <- AddModuleScore_UCell(seurat.object, features = signatures, name = NULL,
ncores = 2)
head(seurat.object@meta.data)
###generate pca and umap embeddings
seurat.object <- NormalizeData(seurat.object)
seurat.object <- FindVariableFeatures(seurat.object, selection.method = "vst", nfeatures = 500)
seurat.object <- ScaleData(seurat.object)
seurat.object <- RunPCA(seurat.object, features = seurat.object@assays$RNA@var.features,
npcs = 20)
seurat.object <- RunUMAP(seurat.object, reduction = "pca", dims = 1:20, seed.use = 123)
FeaturePlot(seurat.object, reduction = "umap", features = names(signatures), ncol = 3,
order = T)
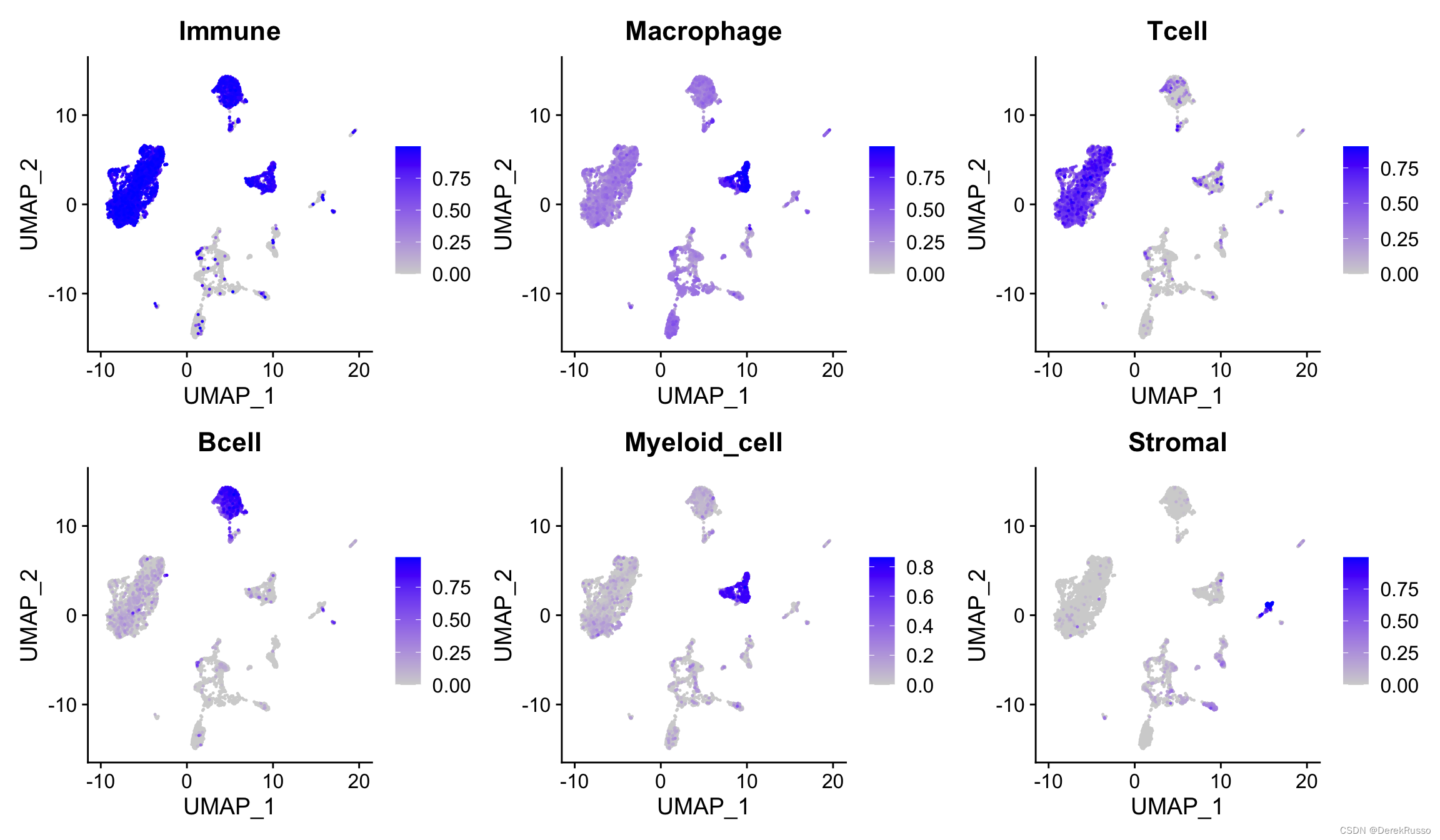
demo3: 完整地展示了seurat标准流程+ucell打分坚定细胞类型+亚群的流程
library(Seurat)
library(UCell)
library(dplyr)
set.seed(123)
inputFile <- "Yost.pretreatment.all.rds"
if (!file.exists(inputFile)) {
download.file("https://drive.switch.ch/index.php/s/cluBLHkFFzLZWzL/download",
inputFile)
}
data.seurat <- readRDS(inputFile)
data.seurat <- data.seurat %>%
NormalizeData %>%
FindVariableFeatures %>%
ScaleData %>%
RunPCA %>%
RunUMAP(dims = 1:30)
data.seurat
### unsupervised clustering
data.seurat <- FindNeighbors(data.seurat, reduction = 'pca', dims = 1:30)
data.seurat <- FindClusters(data.seurat, resolution = 0.7)
DimPlot(data.seurat, reduction = "umap", group.by = "seurat_clusters", label = T) +
NoLegend()
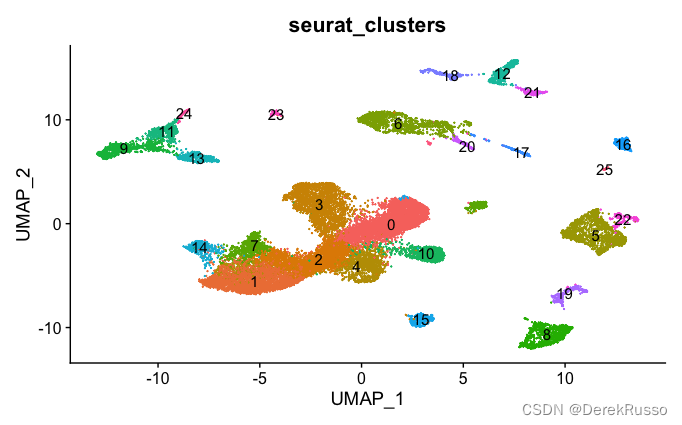
###cell signature
###from https://www.nature.com/articles/s41586-020-2157-4
signaturesHumanCellTypes <- list()
signaturesHumanCellTypes$Fetal.epithelial.progenitor <- c("BEX3" ,"STMN1", "SOX4", "LDHB" , "SKP1", "SNRPE" , "ID3" , "SRP9", "GSTP1" ,"SRP14" )
signaturesHumanCellTypes$Macrophage <- c("CTSB" , "C1QB", "LAPTM5", "TYROBP", "PSAP" , "C1QA", "HLA-DRA", "CTSD" ,"NPC2" , "FCER1G" )
signaturesHumanCellTypes$B.cell..Plasmocyte. <- c("JCHAIN" , "IGHA1" , "SSR4" , "MZB1" , "IGKC" , "IGHA2", "HERPUD1" , "DERL3" , "SEC11C" , "FKBP11" )
signaturesHumanCellTypes$Fibroblast <- c("C1S" , "TIMP2", "COL6A3" ,"SEMA3C" , "MMP2" , "GSN" ,"IGFBP6", "MFAP4", "COL6A1" , "PLAC9")
signaturesHumanCellTypes$Fasciculata.cell <- c("PEBP1" , "STAR", "RARRES2" , "CLU" ,"CYP21A2", "CYP17A1" , "AKR1B1" , "NOV", "TPD52L1" , "EPHX1" )
signaturesHumanCellTypes$T.cell <- c("CD3D" , "CD3E" , "CD3G" , "CD4" , "CD2" , "CD7" , "TRAC", "TRBC1" , "LAT")
### ucell
data.seurat <- AddModuleScore_UCell(data.seurat, features = signaturesHumanCellTypes,
ncores = 2)
# Some major cell types to look at:
toplot <- c("Fetal.epithelial.progenitor", "Macrophage", "B.cell..Plasmocyte.", "Fibroblast", "T.cell", "Fasciculata.cell"
)
featnames <- paste0(toplot, "_UCell")
FeaturePlot(data.seurat, features = featnames, pt.size = 0.1, max.cutoff = "q99",
ncol = 2)
VlnPlot(data.seurat, features = featnames, pt.size = 0, split.by = "seurat_clusters",
ncol = 2)
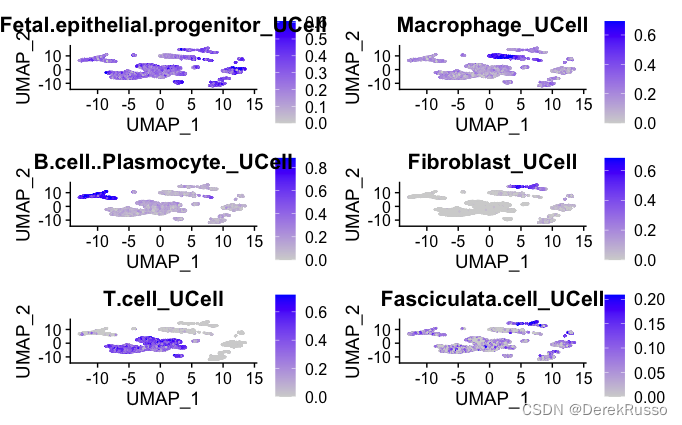
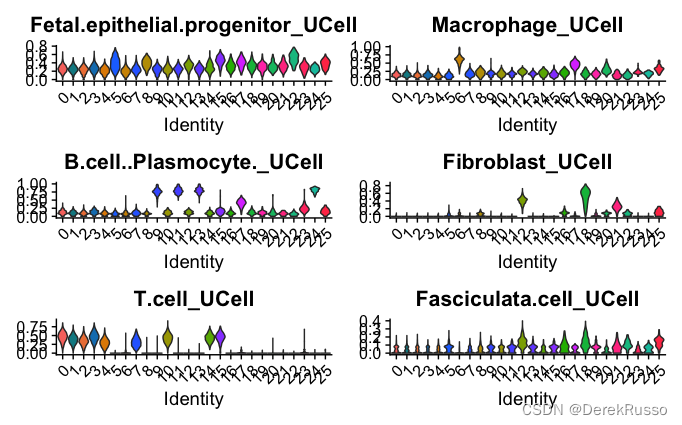
###T.cell_UCell score > 0.2 as threshold
### identify t cells and subset them
# select as Tcell clusters only those with median Uscore>0.2
medians <- sapply(levels(data.seurat$seurat_clusters), function(x) {
median(data.seurat@meta.data[data.seurat$seurat_clusters == x, "T.cell_UCell"])
})
tcell.clusters <- names(medians[medians > 0.2])
# Add metadata
data.seurat$is.Tcell <- FALSE
data.seurat@meta.data[data.seurat$seurat_clusters %in% tcell.clusters, "is.Tcell"] <- TRUE
DimPlot(data.seurat, group.by = "is.Tcell")
#subset on t cells
data.seurat.tcells <- subset(data.seurat, subset = is.Tcell == TRUE)
data.seurat.tcells
#recalculate embedding only for t cells
data.seurat.tcells <- data.seurat.tcells %>%
NormalizeData %>%
FindVariableFeatures %>%
ScaleData %>%
RunPCA %>%
RunUMAP(dims = 1:20)
###Score tumor-infiltrating t cell subtype-specific signatures using UCell
#https://github.com/carmonalab/ProjecTILs
signaturesHumanTILs <- list()
signaturesHumanTILs$Tfh <- c("CD4" , "CD40LG" ,"TOX2" , "MAF" , "CD200" , "BATF" )
signaturesHumanTILs$CD8_NaiveLike <- c("CD8A" , "CD8B" , "CCR7" , "IL7R" , "SELL" , "TCF7" , "S1PR1", "LEF1" )
signaturesHumanTILs$CD8_EffectorMemory <- c("CD8A", "CD8B" , "GZMA" , "GZMK", "CCL5" , "CXCR3")
signaturesHumanTILs$Thelper <- c("CD40LG", "CD4", "IL7R" , "RORA" , "ANXA1" )
signaturesHumanTILs$CD4_NaiveLike <- c("CD40LG" ,"CD4" , "CCR7" , "SELL" , "IL7R" , "TCF7" , "LEF1" )
signaturesHumanTILs$CD8_Tpex <- c("CD8A" , "CD8B" , "LAG3" , "XCL1" , "CRTAM" ,"TOX" , "ZEB2" , "PDCD1" ,"TCF7", "CCR7" )
signaturesHumanTILs$CD8_Tex <- c("CD8A" , "CD8B" , "LAG3" , "HAVCR2", "GZMB" , "PRF1" , "PDCD1" , "TIGIT" )
signaturesHumanTILs$Treg<- c("CD4" , "IL2RA", "FOXP3")
signaturesHumanTILs[["cycling"]] <- c("TOP2A", "MKI67", "STMN1")
data.seurat.tcells <- AddModuleScore_UCell(data.seurat.tcells, features = signaturesHumanTILs,
ncores = 2)
featnames <- paste0(names(signaturesHumanTILs), "_UCell")
FeaturePlot(data.seurat.tcells, features = featnames, pt.size = 0.1, order = T)
VlnPlot(data.seurat.tcells, features = featnames, pt.size = 0, split.by = "seurat_clusters")

















 9734
9734

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









