



概要
本文作为记录贴,记录包括环境配置,数据源下载,原始数据处理,单细胞分析脚本等。
欢迎大佬指导!
环境配置
Windows:
- R 4.5.0
- RStudio 2025.05.1
Linux:
- cellranger-9.0.1
数据源
数据源为国家生物技术信息中心.
以某篇文章为例, 展示查找数据源的过程.
- 找到文章中Data and Code Availability或类似表示数据源的部分, 进入链接.

- 点击下载数据.

- 点击想要下载的数据编号.

- 有两种下载方式, 我这里选择的是HTTPS, 使用迅雷进行下载
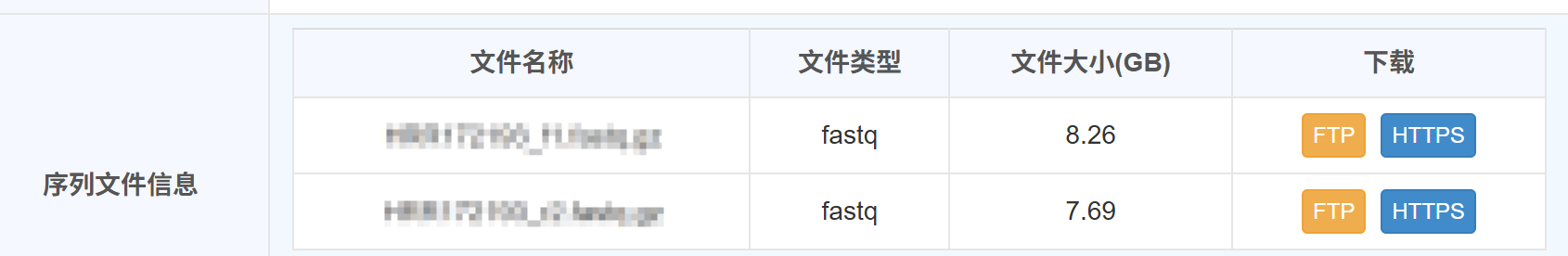
- 下载的原始数据如下
CellRanger处理数据
CellRanger命名格式
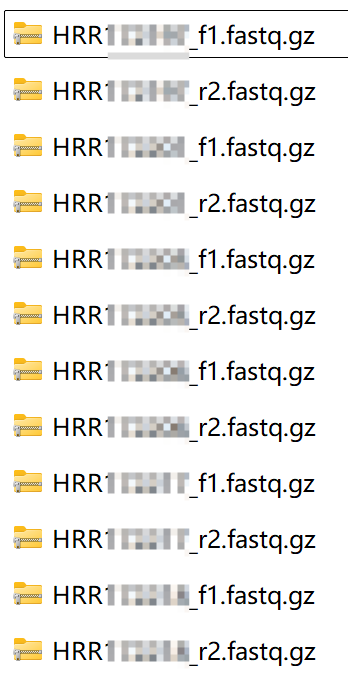
需要通过cellRanger将双端数据(f1, r2)处理为barcodes.tsv.gz, features.tsv.gz, matrix.mtx.gz以进一步分析.
cellRanger有严格的命名规则, 如果直接用HRR*_f1/r2.fastq.gz将会报以下错误.
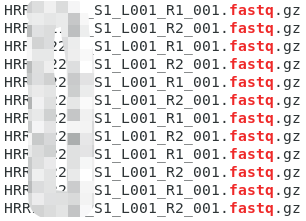
根据提示可知应该将数据命令为SampleName_S1_L001_R1_001.fastq.gz的格式. 即**[Sample Name]_S[Sample Number]_L[Lane Number]_R[Read Type]_001.fastq.gz**.

统一命名后的文件如下, 可以与原始文件的命名进行比较, 将f1改为R1, 将r2改为R2. 统一命令后的文件为:
CellRanger count
由于我们有多组数据需要处理, 批量处理脚本如下, 需要将样本按照samples.csv的格式列出, R1和R2为同一组数据.
#.samples.csv
HRR17xxx7,/raw_data,young_1
HRR17xxx1,/raw_data,young_2
HRR17xxx5,/raw_data,middle_1
HRR17xxx6,/raw_data,middle_2
HRR17xxx7,/raw_data,old_1
HRR17xxx8,/raw_data,old_2
#!bash
while IFS=, read -r sample_id fastq_path age_group
do
cellranger count \
--id=${sample_id}_out \
--transcriptome=$REF \
--fastqs=$fastq_path \
--sample=$sample_id \
--localcores=8 \
--localmem=64 \
--create-bam true
done < samples.csv
cellRanger count处理后的数据为:
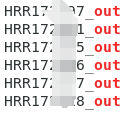
Read10X所需的信息就生成完成啦!
CellRanger aggr
同样的需要一个csv文件将需要aggr的数据列出, 并进行分组, 方便后续处理.
#.aggregation.csv
sample_id,molecule_h5,group,age_group
HRR17xxx7,/raw_data/HRR17xxx7_out/outs/molecule_info.h5,young,young1
HRR17xxx1,/raw_data/HRR17xxx1_out/outs/molecule_info.h5,young,young2
HRR17xxx5,/raw_data/HRR17xxx5_out/outs/molecule_info.h5,middle,middle1
HRR17xxx6,/raw_data/HRR17xxx6_out/outs/molecule_info.h5,middle,middle2
HRR17xxx7,/raw_data/HRR17xxx7_out/outs/molecule_info.h5,old,old1
HRR17xxx8,/raw_data/HRR17xxx8_out/outs/molecule_info.h5,old,old1
#!bash
cellranger aggr \
--id=aggregation_out \
--csv=aggregation.csv \
--normalize=mapped
合并后的数据与单个类似, 包括barcodes.tsv.gz, features.tsv.gz, matrix.mtx.gz, 这里就不放图了.
至此多组数据的fastq已通过cellRanger处理并合并完成, 可以继续进行单细胞分析.
由于cellRanger只能在linux系统使用, 如果后续分析想在windows进行, 可以使用scp命令复制到windows中.
scp -r root@192.xxx.xxx.xxx:/raw_data/HRR_out Y:/scRNA_seq/10x_data
参考
cellranger 操作笔记-1:分析数据命名规则
cellranger使用的初步探索(3)cellranger aggr
scp命令用于Linux和Windows之间传送复制文件
Linux 查看本机 IP 地址命令(超详细)

















 9782
9782

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









