









本期给大家解释介绍如何利用R语言对单个样本的单细胞数据进行分析。
一、单细胞分析流程——单样本
#####加载包#####
library(Seurat)
library(ggplot2)
library(dplyr)
#####00单样本10xGenomics(mtx、.tsv)数据加载及Seurat对象创建######
##加载数据##
remove(list = ls()) #清除Global Environment
getwd() #查看当前工作路径
setwd("D:/Rdata/jc/单细胞演示数据/10xGenomics/") #设置需要的工作路径
list.files() #查看当前工作目录下的文件
seurat.date = Read10X(data.dir = "D:/Rdata/jc/单细胞演示数据/10xGenomics/单样本演示" ) #此步需要去除文件前缀方可正常运行,例如GSM8421890_2021_31_barcodes.tsv.gz,去除前缀变为barcodes.tsv.gz。
##创建seurat对象并初步过滤基因和细胞##
seurat.object = CreateSeuratObject(counts = seurat.date,
project = "GSM8421890_2021_31", #改成相应的样本名
min.features = 200, #每个细胞至少表达的基因数(默认200-500),高质量数据可适当提高数值
min.cells = 3 #每个基因至少在多少个细胞中表达(默认3-10)
)
##查看数据##
seurat.object #查看Seurat对象
dim(seurat.object) # 查看过滤后的基因数和细胞数
head(seurat.object@meta.data) # 查看元数据(默认包含 nCount_RNA, nFeature_RNA)
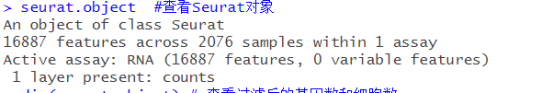
#####001质量控制(QC)#####
##计算线粒体基因比例##
seurat_obj <- PercentageFeatureSet(seurat.object,
pattern = "^MT-", #^MT-人类;^mt-小鼠
col.name = "percent.mt"
)
# pattern = "^RP[SL]")
##可视化QC指标##
VlnPlot(seurat_obj,
features = c("nFeature_RNA", "nCount_RNA", "percent.mt"),
pt.size = 0.1,
ncol = 3
)
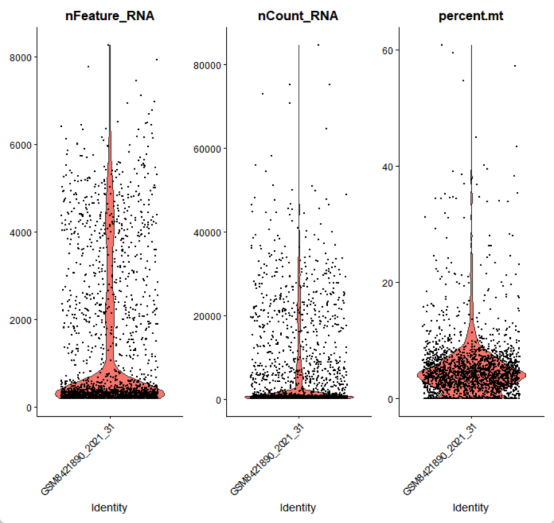
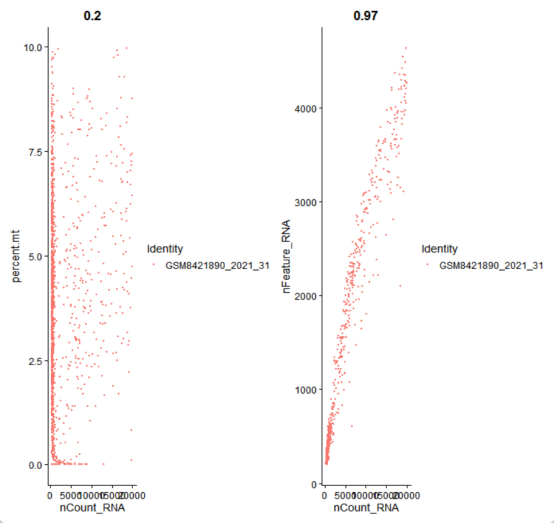
p1 <- FeatureScatter(seurat_obj, #“features”间关系可视化
feature1 = "nCount_RNA",
feature2 = "percent.mt",
pt.size = 0.1
)
p2 <- FeatureScatter(seurat_obj, #“features”间关系可视化
feature1 = "nCount_RNA",
feature2 = "nFeature_RNA",
pt.size = 0.1
)
p3 <- p1+p2
p3
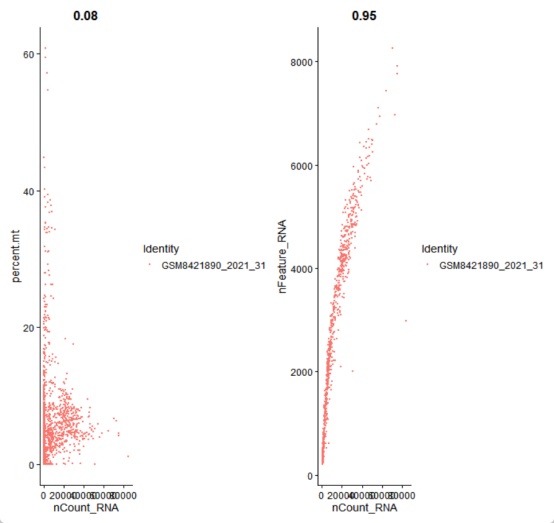
##过滤细胞##
seurat_obj_GL <- subset(seurat_obj,
subset = nFeature_RNA > 200 & #避免低基因数的无效细胞。
nFeature_RNA < 6000 & #避免双细胞或多细胞
nCount_RNA > 300 &
nCount_RNA < 20000 &
percent.mt < 10 #通常设置为5%-10%
)
##检查过滤后结果##
cat("过滤前细胞数:", ncol(seurat_obj), "\n")
cat("过滤后细胞数:", ncol(seurat_obj_GL), "\n")
cat("过滤比例:", round(ncol(seurat_obj_GL)/ncol(seurat_obj), 2), "\n")
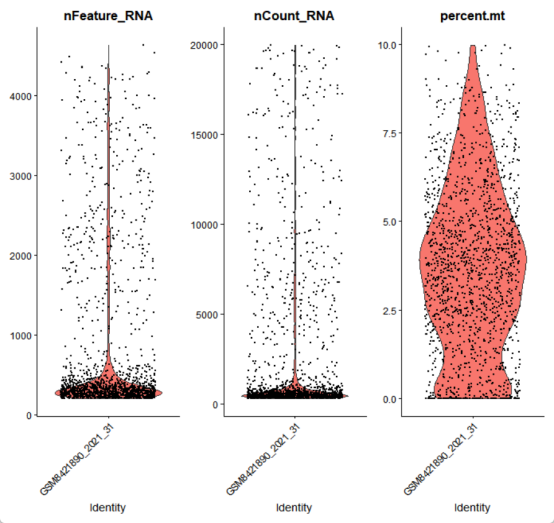
##过滤细胞可视化##
VlnPlot(seurat_obj_GL,
features = c("nFeature_RNA", "nCount_RNA", "percent.mt"),
ncol = 3
)
P4 <- FeatureScatter(seurat_obj_GL, group.by="orig.ident", #“features”间关系可视化
feature1 = "nCount_RNA",feature2 = "percent.mt",pt.size = 0.1)
P5 <- FeatureScatter(seurat_obj_GL, group.by="orig.ident", #“features”间关系可视化
feature1 = "nCount_RNA",feature2 = "nFeature_RNA",pt.size = 0.1)
P <-P4+P5
P
##保存结果,可暂时不执行##
saveRDS(seurat_obj_GL, file = paste(GSM8421890_2021_31,"qc.RDS",sep="."))
数据标准化:校正技术偏差:由于测序深度不同,需标准化基因表达量。
##保存结果,可暂时不执行##
saveRDS(seurat_obj_GL, file = paste(GSM8421890_2021_31,"qc.RDS",sep="."))
#####002标准化与特征选择#####
##数据归一化(标准化)##
seurat_obj_GL <- NormalizeData(seurat_obj_GL, # 对数归一化
normalization.method = "LogNormalize",
scale.factor = 10000)
##高变基因筛选##
seurat_obj_GL <- FindVariableFeatures(seurat_obj_GL,
selection.method = "vst",
nfeatures = 3000) #如果数据稀疏可减少
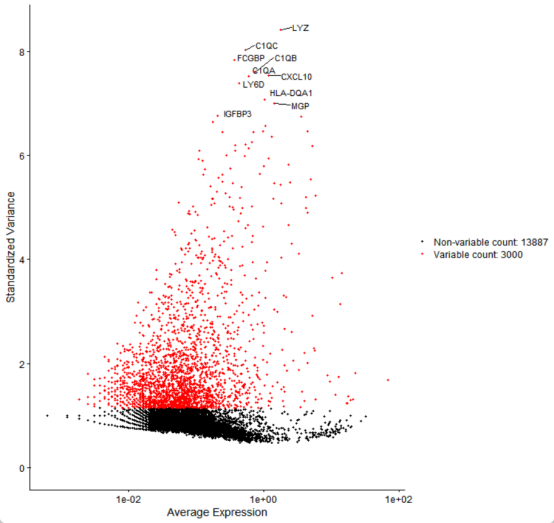
top10 <- head(VariableFeatures(seurat_obj_GL), 10) ##查看找到的前10个高变基因##
print(top10)
#可视化top10高变基因
p4 <- VariableFeaturePlot(seurat_obj_GL)
p5 <- LabelPoints(plot = p4,
points = top10,
repel = TRUE)
p5
##保存高变基因列表(可选)##
write.csv(VariableFeatures(seurat_obj_GL), "highly_variable_genes.csv")
#####003数据缩放与PCA分析#####
##缩放数据##
high.var.genes <- VariableFeatures(seurat_obj_GL) #针对高变基因的缩放
#high.var.genes <- rownames(seurat_obj_GL) #针对选择全部基因的缩放
seurat_obj_GL <- ScaleData(seurat_obj_GL, features = high.var.genes)
##PCA降维##
seurat_obj_GL <- RunPCA(seurat_obj_GL,
features = VariableFeatures(object = seurat_obj_GL)
)
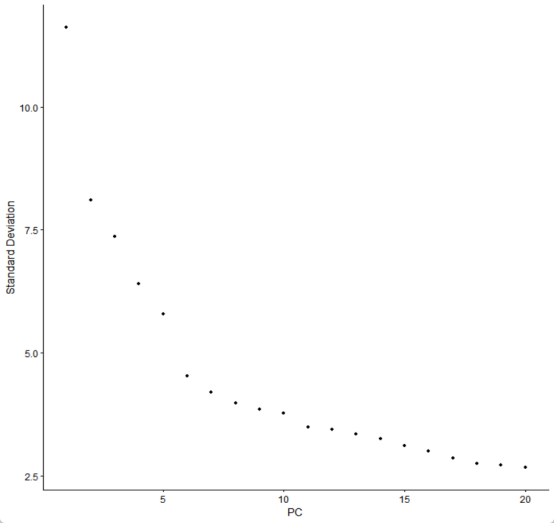
##查看PCA结果##
print(ElbowPlot(seurat_obj_GL)) #确认数据集的维度,选择“肘部”
#可视化PCA降维结果
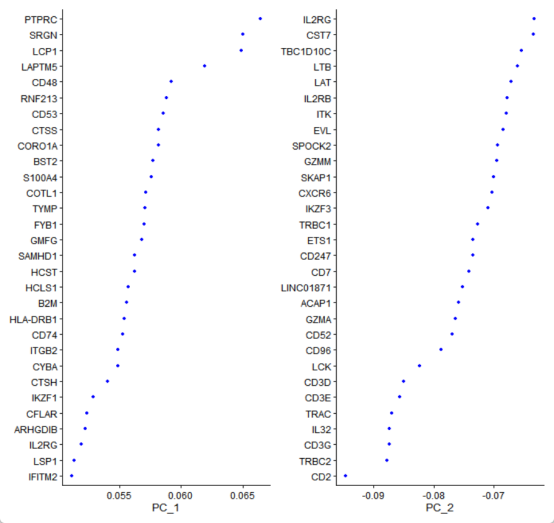
#可视化PCA降维结果
VizDimLoadings(seurat_obj_GL, dims =1:2, reduction ="pca")
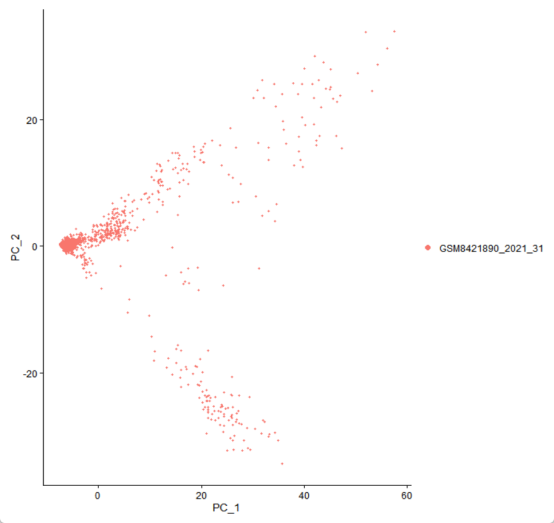
DimPlot(seurat_obj_GL, reduction = "pca") #基于PCA降维的结果绘制散点图
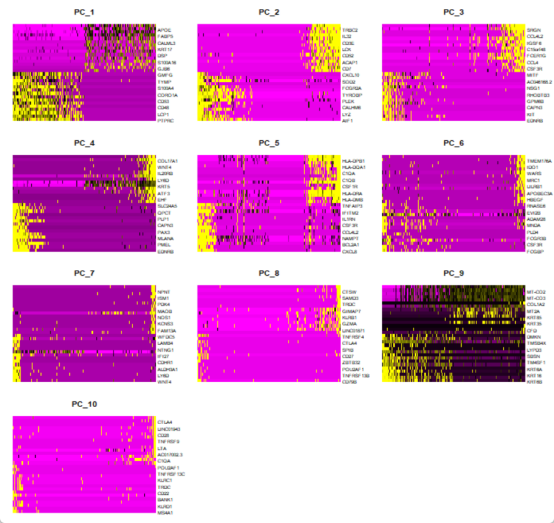
DimHeatmap(seurat_obj_GL, dims =1:10, cells =500, balanced = TRUE) #绘制热图
#####006聚类分析#####
##基于选定的PCs进行聚类##
seurat_obj_GL <- FindNeighbors(seurat_obj_GL,
dims = 1:10) #指定了在计算邻居关系时使用的主成分范围。这里,dims = 1:10意味着将使用前10个主成分来构建最近邻图。
seurat_obj_GL <- FindClusters(seurat_obj_GL,
resolution = 0.5) #根据需要调整resolution参数
head(Idents(seurat_obj_GL), 5) #请求只查看前5个细胞的身份标识。
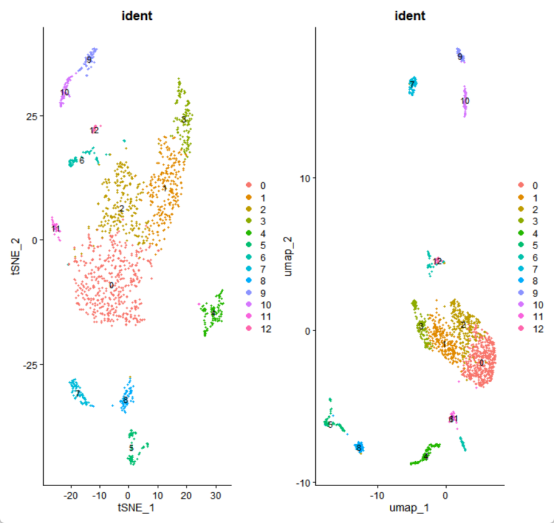
##运行UMAP和tSNE##
#tSNE降维及可视化
seurat_obj_GL <- RunTSNE(seurat_obj_GL, dims = 1:10) #dims根据PCA的结果来决定使用多少个主成分。
p7 <- DimPlot(seurat_obj_GL,
reduction = "tsne",
group.by = "ident", #指定按照哪个元数据变量对点进行分组着色
label = TRUE #这个参数会在图上显示群集标签
)
#UMAP降维及可视化
seurat_obj_GL<- RunUMAP(seurat_obj_GL, dims = 1:10)
p8 <-DimPlot(seurat_obj_GL,
reduction = "umap",
group.by = "ident",
label = TRUE
)
p9<- p7 + p8
p9
#####007鉴定细胞簇间的差异表达基因#####
# 每个细胞簇找对比其他所有细胞簇的差异基因(1)
seurat_obj_markers <- FindAllMarkers(seurat_obj_GL, #直接按参数进行筛选全部cluster间差异
only.pos = F, #保留所有显著差异表达的基因;TRUE(只保留上调基因)
min.pct = 0.25, # 至少在25%的细胞中表达该基因才会被考虑。
logfc.threshold = 1
)
diff.markers <- seurat_obj_markers %>% filter(p_val_adj < 0.05) #筛选差异显著的基因
print(seurat_obj_markers)
# 对每个细胞簇找对比其他所有细胞簇的差异基因(2)#
#seurat_obj.all.markers <- FindAllMarkers(seurat_obj_GL, only.pos = F) #所有簇间的差异基因
#seurat_obj.markers <- seurat_obj.all.markers %>% #根据参数再进行筛选
#group_by(cluster) %>%
#dplyr::filter(abs(avg_log2FC) > 1)
#diff.markers <- seurat_obj.markers %>% filter(p_val_adj < 0.05) #筛选差异显著的基因
#print(seurat_obj.markers)
#识别cluster2相对于其他所有簇的特异性上调基因,但是roc,wilcox更适合区分两个群体#
cluster2.markers <- FindMarkers(seurat_obj_GL,
ident.1 = 2,
logfc.threshold = 1,
#test.use = "roc", #更适合寻找那些特异性高但 fold change 较小的 marker 基因,特别是在数据集不平衡的情况下
#test.use = "wilcox", #则更适合寻找表达差异显著的 marker 基因,并且能够提供统计显著性度量。
only.pos = TRUE #上调
)
#合并"roc"和"wilcox"结果看总体结果
#combined_markers <- merge(markers_roc, markers_wilcox, by = "gene", suffixes = c("_roc", "_wilcox"))
head(cluster2.markers, n = 5)
#找细胞簇5对比细胞簇0和3的差异基因#
cluster5.markers <- FindMarkers(seurat_obj_GL,
ident.1 = 5,
ident.2 = c(0, 3)
)
head(cluster5.markers, n = 5)
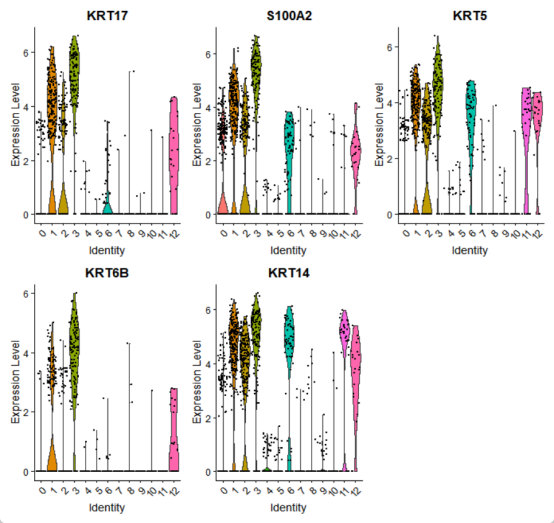
##可视化基因表达##
#小提琴图
VlnPlot(seurat_obj_GL,
features = c("KRT17","S100A2","KRT5","KRT6B","KRT14")
)
#基因的细胞聚类分布图
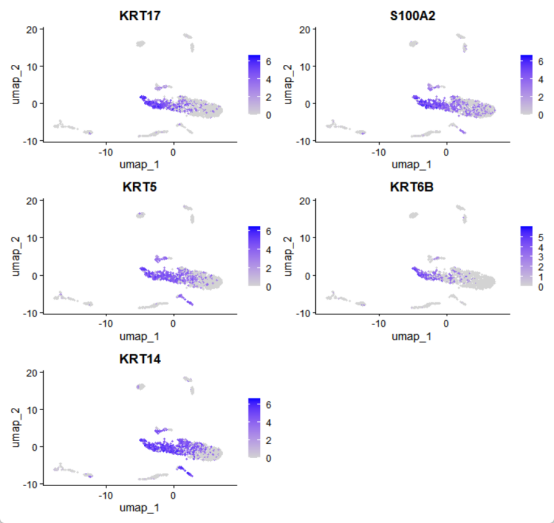
FeaturePlot(seurat_obj_GL,
features = c("KRT17","S100A2","KRT5","KRT6B","KRT14")
)
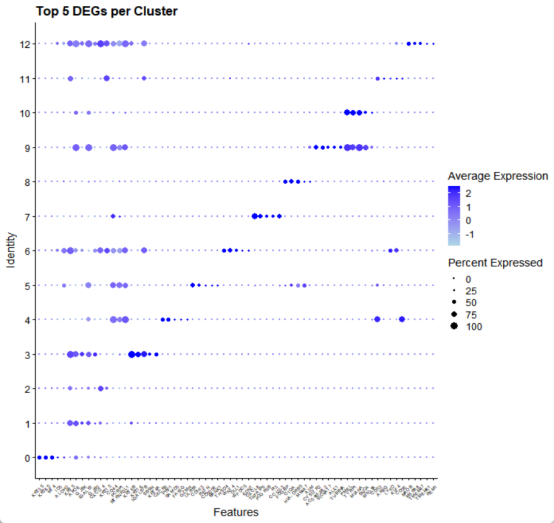
#查看top5DEGs的表达气泡图
top5 = seurat_obj_markers %>%
group_by(cluster) %>%top_n(n = 5, wt = avg_log2FC) #需要包含所有基因及其在不同细胞簇间的表达差异信息的数据框
p <- DotPlot(
seurat_obj_GL,
features = top5$gene,
cols = c("lightblue", "blue"), # 设置颜色渐变
dot.scale = 4 # 调整点的大小(默认为6)
) + RotatedAxis() + # 旋转y轴标签
theme(
panel.background = element_rect(fill = "white"),
plot.background = element_rect(fill = "white"),
axis.text.y = element_text(size = 12, color = "black"), # 调整y轴标签字体大小和颜色
axis.text.x = element_text(size = 6, color = "black") # 调整x轴标签字体大小和颜色
) +
labs(title = "Top 5 DEGs per Cluster") # 添加标题
p
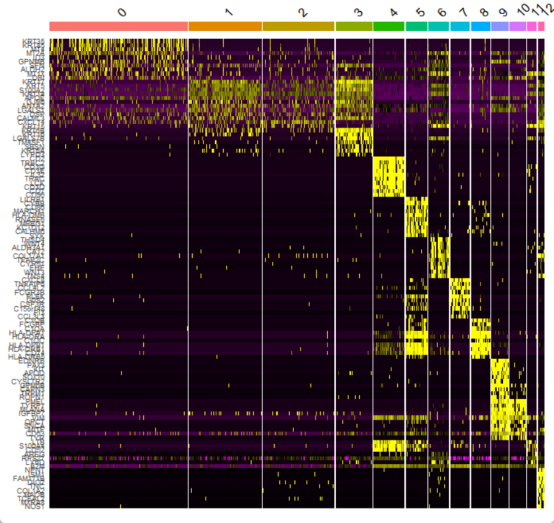
#热图
seurat_obj_markers %>%
group_by(cluster) %>% #按照cluster列分组,即每个簇单独处理
dplyr::filter(avg_log2FC > 1) %>% #筛选出在该簇中log2 fold change > 1 的基因(即显著上调的基因)
slice_head(n = 10) %>% #对每个簇保留前10个差异最显著的基因(按默认排序,通常是按log2FC从高到低)
ungroup() -> top10 #取消分组,恢复数据框为普通格式。将最终结果保存到变量 top10 中,它是一个包含每个簇前10个显著上调基因的列表。
DoHeatmap(seurat_obj_GL, features = top10$gene) + NoLegend()
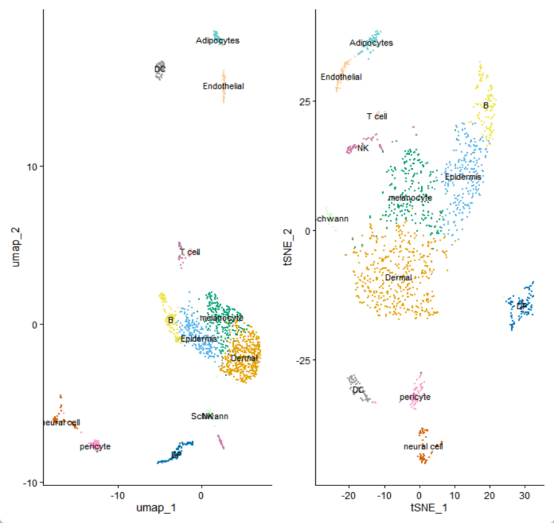
#####008细胞类型注释#####
#已知各类型细胞后进行注释#
new.cluster.ids <- c("Dermal","Epidermis","melanocyte","B","DP","neural cell", "NK", "DC", "pericyte","Adipocytes","Endothelial","Schwann","T cell")
names(new.cluster.ids) <- levels(seurat_obj_GL)
seurat_obj_GL <- RenameIdents(seurat_obj_GL, new.cluster.ids)
my_colors <- c("#E69F00", "#56B4E9", "#009E73", "#F0E442", "#0072B2",#配色方案可根据多少个cluster而后去AI找
"#D55E00", "#CC79A7", "#999999", "#FF99CC", "#66CCCC",
"#FFCC99", "#CCFFCC", "#FFCCCC")
p10 <- DimPlot(seurat_obj_GL, #可视化#
reduction = "umap",
label = TRUE,
pt.size = 0.5,
cols = my_colors) + NoLegend()
p11 <- DimPlot(seurat_obj_GL, #可视化#
reduction = "tsne",
label = TRUE,
pt.size = 0.5,
cols = my_colors) + NoLegend()
p12 <- p10+p11
p12
好了本次分享就到这里,下期将有更精彩单细胞分析内容,敬请期待。
关注”在打豆豆的小潘学长“公众号,还有更精彩内容。

















 1万+
1万+

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









