







PoseBusters: AI-based docking methods fail to generate physically valid poses or generalise to novel sequences†
摘要:
近年来,基于深度学习的蛋白质-配体对接方法得到了广泛发展。这些方法在速度和准确性方面展现出了巨大潜力。然而,尽管在结晶学根均方差(RMSD)方面宣称具有最先进的性能,但经过仔细检查后,明显发现它们往往产生了不符合物理规律的分子结构。因此,单单通过与原生结合模式的RMSD来评估这些方法是不够的。特别是对于基于深度学习的方法,评估时必须考虑到立体化学和能量学等标准。为此,我们提出了PoseBusters,这是一个基于Python的工具包,能够通过使用成熟的化学信息学工具包RDKit进行一系列标准质量检查。PoseBusters的测试套件验证了配体的化学和几何一致性,包括其立体化学,以及分子内部和分子间的物理合理性,例如芳香环的平面性、标准键长以及蛋白质-配体之间的碰撞等。只有那些通过这些检查并预测出与原生结合模式相似的结合模式的方法,才能被视为具有“最先进”性能的。我们使用PoseBusters对五种基于深度学习的对接方法(DeepDock、DiffDock、EquiBind、TankBind和Uni-Mol)以及两种成熟的标准对接方法(AutoDock Vina和CCDC Gold)进行了比较,并在添加了分子力学能量最小化步骤(使用分子力学力场)后再次比较。我们发现,在物理合理性和对与训练数据不同的示例进行泛化的能力方面,目前没有任何基于深度学习的方法超越传统的对接工具。此外,我们发现分子力学力场包含了深度学习方法中缺失的对接相关的物理特性。PoseBusters使实践者能够评估对接和分子生成方法,并可能激发新的归纳偏置,进一步改善基于深度学习的方法,推动更准确和更具现实性的预测的发展。
介绍:
对接是结构基础药物发现中的一个关键步骤,它的任务是预测蛋白质-配体复合物的主要结合模式,前提是已知实验解析或计算模型得到的蛋白质结构和配体结构。预测的复合物通常用于虚拟筛选流程,以帮助从大量候选分子中选择分子;或被药物化学家直接用于理解结合模式,并决定某一小分子是否是合适的药物候选者。对接方法的设计是基于这样一个认识:结合是通过靶标和配体结构之间的相互作用实现的,但由于这一特性的复杂性,这些方法通常在计算速度和准确性之间做出平衡。深度学习(DL)有可能颠覆传统对接软件的主导设计原则,而基于深度学习的对接方法则有望为药物发现提供快速且准确的虚拟筛选。为此,已经提出了一些不同的基于深度学习的对接方法。传统的非深度学习(non-DL)对接方法在其搜索和评分函数中包含一些有助于确保化学一致性和物理合理性的项。例如,它们通过限制配体的运动范围仅限于配体的可旋转键,并在蛋白质和配体发生碰撞时加以惩罚。正如我们所展示的,一些当前的基于深度学习的对接方法仍然缺乏这些关键的“归纳偏置”,尽管它们能获得低于广泛使用的2Å阈值的实验结合模式根均方差(RMSD)值,但仍然会产生不现实的构象。为了评估这些对接方法,需要一个独立的测试套件来检查化学一致性和物理合理性,并结合已建立的评估标准,如结合模式的RMSD值。这样的测试套件将帮助领域内识别缺失的归纳偏置,从而改进基于深度学习的对接方法,推动更准确和更现实的对接预测的发展。评估对接预测的物理合理性的问题类似于对蛋白质数据银行(PDB)中配体数据的结构验证。结构验证评估配体的键长和键角与相关化学结构中观察到的值的一致性,以及配体内部和配体与其周围环境之间的立体冲突。尽管这些测试是为了帮助用户选择那些可能是正确的配体晶体结构,但对接方法的评估则侧重于它们恢复晶体结构的能力,因此它们的输出也应该通过相同的物理合理性测试。物理合理性检查也是一些构象生成工作流程的一部分。Friedrich 等人使用了由 NAOMI 执行的几何检查,该检查类似于上述 PDB 测试,测量了键长和键角偏离已知最优值的程度,并测试了芳香环平面性的偏离。除了物理检查,化学检查也是必要的。为检查 PDB 结构提出的化学检查包括识别标注错误的立体化学分配、不一致的键合模式、缺失的功能团和不太可能的离子化状态。检查化学合理性的问题在从头分子生成中也出现过,Brown 等人提出了一个测试套件,包括检查任何提出的分子化学有效性的内容。对于对接而言,关注点较少在分子结构的稳定性和合成可达性上,因为期望在进行对接之前这些问题已被测试过,更多的是在于预测结合构象的化学一致性和物理现实性。一些对接方法的比较已经包括了基于体积重叠或蛋白质-配体相互作用的附加指标,以补充基于构象准确性的指标,如原子位置的RMSD和运行时间测量,但大多数对接方法的比较仍然主要基于结合模式的RMSD。当前通过仅基于RMSD指标比较对接方法的标准做法也延续到最近新方法的介绍论文中。我们在本文中测试的五种基于深度学习的对接方法都声称其性能优于标准对接方法,但这些声明完全基于RMSD。这些方法没有对其输出进行物理合理性测试。在本文中,我们介绍了PoseBusters,这是一个旨在识别不合理构象和配体姿势的测试套件。我们使用PoseBusters评估了由五种基于深度学习的对接方法(DeepDock、DiffDock、EquiBind、TankBind和Uni-Mol)以及两种标准的非深度学习对接方法(AutoDock Vina和Gold)生成的预测配体姿势。这些姿势是通过将81个蛋白质-配体晶体复合物(来自Astex Diverse数据集)和308个蛋白质-配体晶体复合物(来自PoseBusters基准集,一组从2021年开始发布的新复合物)的配体重新对接到其对应的受体晶体结构中生成的。在广泛使用的Astex Diverse数据集上,基于深度学习的对接方法DiffDock在仅考虑RMSD时似乎表现最佳,但在考虑物理合理性时,Gold和AutoDock Vina表现最佳。在PoseBusters基准集上,这是一组更具挑战性的测试集,因为它仅包含深度学习方法未经过训练的复合物,Gold和AutoDock Vina在仅考虑RMSD时表现最佳,并且在考虑物理合理性或考虑具有新序列的蛋白质时也表现最佳。基于深度学习的方法在未见过的复合物上做出的有效预测很少。总体来说,我们表明,在考虑物理合理性时,基于深度学习的方法尚未超越标准的对接方法。PoseBusters测试套件将使深度学习方法的开发者更好地理解当前方法的局限性,最终推动更准确和更现实的预测的产生。
方法:
我们使用了五种基于深度学习的对接方法和两种经典的对接方法,将已知配体重新对接到它们各自的蛋白质中,并使用PoseBusters测试套件评估预测的配体姿势。接下来的部分将描述对接方法、数据集以及PoseBusters测试套件,用于检查生成的配体姿势的物理化学一致性和结构合理性。
对接方法:
所选的五种基于深度学习的对接方法涵盖了广泛的深度学习方法用于姿势预测。为了检验标准非深度学习对接方法预测化学和物理有效姿势的能力,我们还包括了两个成熟的对接方法:AutoDock Vina和Gold。这五种基于深度学习的对接方法可以总结如下,DeepDock通过学习基于配体重原子与结合口袋表面网格点之间距离的统计势能来进行对接。DiffDock使用等变图神经网络和扩散过程进行盲对接。EquiBind使用等变图神经网络进行盲对接。TankBind是一种盲对接方法,使用一个三角函数感知的神经网络,在由结合口袋预测方法预测的每个口袋中进行对接。Uni-Mol使用SE3等变换器进行对接。所有五种基于深度学习的对接方法都在PDBbind General Set的子集上进行了训练,DeepDock是基于v2019训练的,其余四种方法则是基于v2020训练的。需要注意的是,我们使用了各方法作者训练的深度学习模型,未进行进一步的超参数调优。用于每种方法生成预测的对接协议以及使用的软件版本在补充信息的第S1节中提供。表3列出了我们为每种方法定义的搜索空间。DeepDock和Uni-Mol需要定义一个结合位点,而DiffDock、EquiBind和TankBind是“盲”对接方法,在整个蛋白质范围内搜索。对于DiffDock、DeepDock、EquiBind和TankBind,我们使用了默认的搜索空间,而对于AutoDock Vina、Gold和Uni-Mol,我们使用了比默认值更大的搜索空间,以便与盲对接方法更具可比性。补充信息中的图S1显示了一个示例蛋白质-配体复合物的搜索空间。我们展示了Uni-Mol在多种结合位点定义范围下的结果,从其推荐的紧凑定义开始,即所有距离晶体配体的重原子在6Å范围内的残基。在这种紧凑的口袋定义下,Uni-Mol的表现优于任何盲对接方法(见补充信息图S21)。

The PoseBusters test suite
PoseBusters测试套件分为三个测试组。第一组检查化学有效性,包含相对于输入的化学有效性和一致性测试。第二组检查分子内部特性,测试配体的几何结构和使用通用力场(UFF)计算的配体构象的能量。第三组考虑分子间相互作用,检查蛋白质-配体和配体-辅助因子的碰撞。PoseBusters在这三个部分执行的所有测试的描述列在表4中。通过PoseBusters所有测试的分子姿势被认为是“PB有效的”。 为了评估对接预测,PoseBusters需要三个输入文件:一个包含重新对接配体的SDF文件,一个包含真实配体的SDF文件,以及一个包含蛋白质及其辅助因子的PDB文件。这三个文件会被加载到RDKit分子对象中,并关闭了标准化选项。
表4:测试套件中所用检查描述
| 测试名称 | 描述 |
| 化学有效性和一致性(Chemical validity and consistency) | |
| 文件加载(File loads) | - 描述:输入分子可以通过RDKit被加载到一个分子对象中。 - 解释:这意味着RDKit软件能够正确读取并处理输入的分子数据,将其转化为内部可操作的分子对象格式。 |
| 净化处理(Sanitisation) | - 描述:输入分子通过RDKit的化学净化检查。 - 解释:RDKit会对分子进行一系列检查,确保其符合化学规则,比如原子价态合理、化学键连接符合化学常识等。 |
| 分子式(Molecular formula) | - 描述:输入分子的分子式与真实分子的分子式相同。 - 解释:从元素组成和各元素原子数量比例的角度,验证输入分子化学成分的准确性。 |
| 化学键(Bonds) | - 描述:输入分子中的化学键与真实分子中的化学键相同。 - 解释:包括化学键类型(单键、双键、三键等)、连接的原子种类和化学键在分子结构中的位置都要与真实分子一致,确保分子结构骨架正确。 |
| 四面体手性(Tetrahedral chirality) | - 描述:输入分子中指定的四面体手性与真实分子中的相同。 - 解释:对于具有四面体构型且存在手性中心的分子,手性会影响分子的化学性质和反应活性,所以要确保手性匹配。 |
| 双键立体化学(Double bond stereochemistry) | - 描述:输入分子中指定的双键立体化学与真实分子中的相同。 - 解释:双键的立体化学涉及双键周围原子或基团的空间排列方式,不同的立体化学构型会导致分子具有不同的物理和化学性质,需保证其准确性。 |
| 分子内有效性(Intramolecular validity) | |
| 键长(Bond lengths) | - 描述:输入分子中的键长在由距离几何方法确定的下限的0.75倍到上限的1.25倍范围内。 - 解释:距离几何方法可计算出合理的键长范围,限定在此范围内可保证分子结构的合理性,因为键长异常会影响分子的稳定性和反应性等。 |
| 键角(Bond angles) | - 描述:输入分子中的键角在由距离几何方法确定的下限的0.75倍到上限的1.25倍范围内。 - 解释:键角是分子结构的重要参数,合理的键角范围能维持分子的正常几何构型,确保其化学和物理性质相对稳定。 |
| 平面芳香环(Planar aromatic rings) | - 描述:具有5或6个成员的芳香环中的所有原子在距离最近的共享平面0.25 Å以内。 - 解释:芳香环通常具有平面结构,这种平面性对于芳香性的体现以及分子的电子结构很重要,确保原子在规定距离内接近共享平面可维持芳香环的正常特性。 |
| 平面双键(Planar double bonds) | - 描述:脂肪族碳 - 碳双键的两个碳原子及其四个相邻原子在距离最近的共享平面0.25 Å以内。 - 解释:对于平面双键结构,相关原子在空间上接近共享平面有助于维持双键的平面特性,进而影响分子的立体结构和相关化学性质。 |
| 内部空间位阻冲突(Internal steric clash) | - 描述:非共价键结合的原子对之间的原子间距离高于由距离几何方法确定的下限的0.8倍。 - 解释:空间位阻冲突会影响分子的构象稳定性和分子间的相互作用等,设定此距离标准可避免分子内出现不合理的原子拥挤情况,保证分子结构的合理性。 |
| 能量比(Energy ratio) | - 描述:输入分子的计算能量不超过为输入分子生成的50个构象集合的平均能量的100倍。这里先使用ETKDGv3生成输入分子的50个构象,然后用UFF32在RDKit中计算能量,并且对这些构象还进行了最多200次迭代的力场松弛处理。 - 解释:从能量角度评估输入分子构象的合理性,能量过高或过低可能意味着分子构象存在问题。 |
| 分子间有效性(Intermolecular validity) | |
| 最小蛋白质 - 配体距离(Minimum protein - ligand distance) | - 描述:蛋白质 - 配体原子对之间的距离大于原子对范德华半径之和的0.75倍。 - 解释:确保蛋白质和配体之间在空间上有合适的距离,避免过于靠近产生不合理的相互作用或影响彼此的正常功能。 |
| 到有机辅因子的最小距离(Minimum distance to organic cofactors) | - 描述:配体和有机辅因子原子之间的距离大于原子对范德华半径之和的0.75倍。 - 解释:保证配体与有机辅因子在空间上保持合理距离,防止相互干扰或产生异常的相互作用。 |
| 到无机辅因子的最小距离(Minimum distance to inorganic cofactors) | - 描述:配体和无机辅因子原子之间的距离大于原子对共价半径之和的0.75倍。 - 解释:确保配体与无机辅因子之间有合适的距离,维持它们各自的功能以及合理的相互作用关系。 |
| 与蛋白质的体积重叠(Volume overlap with protein) | - 描述:与蛋白质相交的配体体积份额小于7.5%。这里通过以重原子周围按0.8倍缩放的范德华半径来定义体积。 - 解释:避免配体与蛋白质在空间上过度重叠而影响彼此的功能或产生不合理的相互作用等。 |
| 与有机辅因子的体积重叠(Volume overlap with organic cofactors) | - 描述:与有机辅因子相交的配体体积份额小于7.5%。这里以重原子周围按0.8倍缩放的范德华半径来定义体积。 - 解释:控制配体与有机辅因子在空间上的重叠情况,保证它们之间合理的相互作用关系。 |
| 与无机辅因子的体积重叠(Volume overlap with inorganic cofactors) | - 描述:与无机辅因子相交的配体体积份额小于7.5%。这里以重原子周围按0.5倍缩放的范德华半径来定义体积。 - 解释:维持配体与无机辅因子之间合理的相互作用关系,避免过度重叠带来的不良影响。 |
化学有效性和一致性 (Chemical validity and consistency)
PoseBusters中的第一个测试检查配体是否通过RDKit的标准化处理。RDKit的标准化过程处理分子的价态、芳香性、自由基、共轭、杂化、立体化学标签和质子化信息,以检查一个分子是否能作为八电子完整的路易斯点结构表示。通过RDKit的标准化处理是化学有效性测试中常用的一种方法,特别是在化学信息学中,比如在从头分子生成中使用。PoseBusters中的下一个测试检查预测配体与真实配体之间的对接相关化学一致性。具体做法是生成输入和输出配体的“标准InChI”字符串,过程包括去除同位素信息并通过添加或去除氢原子来中和电荷(在可能的情况下)。InChI是分子比较的事实标准,生成的“标准InChI”字符串包括分子式层(/)、分子键(/c)、氢原子(/h)、净电荷(/q)、质子(/p)、四面体立体化学(/t)和双键立体化学(/b)等层。配体的质子化和电荷状态的标准化是必要的,因为立体化学层依赖于氢原子(/h)、净电荷(/q)和质子(/p)层。这些信息在对接过程中可能会发生意外变化,尽管大多数对接软件将配体的电荷分布和质子化状态视为固定的。标准化协议还会移除初级酮胺中双键的立体化学信息,因为这种信息仅依赖于氢原子的模糊位置。
分子内有效性 (Intramolecular validity)
PoseBusters测试套件中的第一组物理合理性测试验证了对接配体中非共价结合的原子对之间的键长、键角和内部距离,并将其与从RDKit的距离几何模块获得的距离边界矩阵中的相应限制进行比较。为了通过测试,所有分子测量值必须在用户指定的容差范围内。本文中使用的容差为:键长和键角的容差为25%,非共价结合的原子对的容差为30%。例如,如果某个键的长度小于距离几何学给定的键长下限的75%,则被视为异常。这一阈值的选择是因为在Astex Diverse数据集中的所有晶体配体(除一个外)以及PoseBusters基准集中的所有配体在此阈值下都能通过测试。PoseBusters的平面度测试检查一组原子是否位于同一平面上,方法是计算最接近这些原子的平面,并检查所有原子是否在此平面与用户定义的距离范围内。该测试适用于5和6成员的芳香环以及非环状的非芳香性碳-碳双键。选择的阈值为0.25Å,所有Astex Diverse和PoseBusters基准集的晶体结构都能大大通过此阈值,并且与所有其他阈值一样,用户可以根据需要进行调整。PoseBusters进行的最后一个分子内部物理化学合理性测试是能量计算,用于检测不太可能的构象。我们使用的度量标准是对接配体构象的能量与50个生成的无约束构象的能量均值之比,如Wills等人所述。构象是使用RDKit的ETKDGv3构象生成器生成的,然后通过UFF力场进行放松,最多进行200次迭代。测试套件会拒绝那些此比值大于用户指定阈值的构象。Wills等人根据PDBbind数据集中95%的晶体配体被认为是合理的经验值,设定了一个比值为7的阈值。我们选择了一个较宽松的比值为100的阈值,在此阈值下,只有Astex Diverse和PoseBusters基准集中各有一个结构被拒绝。
分子间有效性 (Intermolecular validity)
在PoseBusters测试套件中,分子间相互作用通过两组测试进行评估。第一组测试检查分子之间的最小距离,第二组测试检查重叠体积的比例。两组测试都报告配体与四种分子类型的分子间相互作用:蛋白质、有机辅因子和无机辅因子。对于基于距离的分子间测试,PoseBusters计算两分子重原子对之间的成对距离与这两个原子范德华半径之和的比值。如果该比值小于用户定义的阈值,则测试失败。默认的阈值是0.75,适用于所有分子对。对于无机辅因子-配体对,使用的是共价半径。Astex Diverse数据集中的所有晶体结构以及PoseBusters基准集中的所有晶体结构(除一个外)都能通过此阈值测试。对于第二组分子间检查,PoseBusters使用RDKit的ShapeTverskyIndex函数计算配体重原子范德华体积与蛋白质重原子范德华体积的重叠比例。该测试具有可配置的缩放因子,用于定义范德华半径的体积,并且设置了一个阈值来定义多少重叠构成碰撞。设置阈值是必要的,因为许多晶体结构中本身就已经包含了碰撞。例如,Verdonk等人发现,从PDB中选择的305个高质量蛋白质-配体复合物中,有81个包含立体碰撞。所有分子对的重叠阈值为7.5%,蛋白质-配体和有机辅因子-配体对的缩放因子为0.8,无机辅因子-配体对的缩放因子为0.5。
拟合质量(Quality of Fit)
PoseBusters计算预测的配体结合模式与最接近的晶体配体之间的最小重原子对称根均方差(RMSD),使用的是RDKit的GetBestRMS函数。覆盖度(Coverage)是测试对接方法时常用的一个指标,它表示在用户可调的阈值内的预测占所有预测的比例,默认阈值为2Å的RMSD。这个值是任意选择的,但通常使用并推荐用于常规大小的配体。
序列同一性(Sequence Identity)
在本文中,两个氨基酸链之间的序列同一性是指在序列比对后,两个链中完全匹配的氨基酸残基的数量,除以查询序列中的残基总数。所使用的序列比对方法是Smith–Waterman算法,该算法在Biopython中实现,使用开口缺口得分为-11,延伸缺口得分为-1,并采用BLOSUM62替代矩阵。未知的氨基酸残基被视为不匹配。
分子力学能量最小化
在对接后,对结合口袋中的配体结构进行能量最小化,使用的是AMBER ff14sb力场和Sage小分子力场,在OpenMM中进行。蛋白质文件是通过PDBfixer准备的,所有蛋白质原子的位置在空间中是固定的,只允许更新配体原子的位置。最小化过程一直进行,直到能量收敛至0.01 kJ/mol。
Data
Astex Diverse 数据集
Astex Diverse 数据集(发表于2007年)是从PDB中精心挑选的、相关的、多样的和高质量的蛋白质-配体复合物集合。这些复合物从PDB下载为MMTF文件,并使用PyMOL软件去除溶剂和所有感兴趣配体的出现,然后将蛋白质(包括辅因子)保存为PDB文件,配体则保存为SDF文件。
PoseBusters基准集
PoseBusters基准集是一个新的、经过精心挑选的来自PDB的公开晶体复合物集合。它是一个多样化的、包含药物样分子的近期高质量蛋白质-配体复合物集合。该数据集仅包含自2021年发布的复合物,因此不包含用于训练许多方法的PDBbind General Set v2020中的复合物。表S2列出了选择PoseBusters基准集中308个独特蛋白质和308个独特配体的步骤。这些复合物从PDB下载为MMTF文件,使用PyMOL去除溶剂和所有感兴趣配体的出现,然后将蛋白质(包括辅因子)保存为PDB文件,配体保存为SDF文件。
结果:
以下部分展示了PoseBusters测试套件对五种基于深度学习的对接方法和两种标准非深度学习对接方法进行评估的结果,这些方法在Astex Diverse数据集的85个配体和PoseBusters基准集的308个配体上进行重新对接,配体被对接到对应的受体晶体结构中。
数据可用性
PoseBusters作为一个可通过pip安装的Python包,以及开源代码,按照BSD-3-Clause许可证发布,代码托管在GitHub上,地址为:https://github.com/maabuu/posebusters。本文的数据,包括Astex Diverse数据集和PoseBusters基准集,以及每个对接的单独测试结果,均可在Zenodo平台上访问,链接为:https://zenodo.org/records/8278563。
附加
PoseBusters的理解与使用
PoseBusters基准集中有428条数据,每一条数据都有三个sdf文件和一个pdb文件保存在一个文件夹下
-
xxx_ligand.sdf(参考配体结构):- 这个文件通常包含配体的 标准结构,是实验验证的、经过优化或最优构象的配体结构。它代表了配体在正常条件下的典型状态或通过实验得到的最优构象。
-
xxx_ligand_start_conf.sdf(配体的初始构象):- 这个文件包含配体的 初始构象,通常是通过对接或其他预测方法生成的初步构象。它可能并不完全符合配体的最优构象,而是对接或建模过程中的一个开始状态,可能包含一些不太优化的位姿。
-
xxx_ligands.sdf(多个配体位姿):- 这个文件通常包含多个配体的不同构象(位姿),这些构象是通过对接算法生成的,可以是同一配体在不同结合位点或不同对接结果下的位姿。
xxx_protein.pdb- 这个文件描述了一个蛋白质的三维结构,包含了蛋白质的氨基酸链和通过
HETATM标记的配体信息。假设这个蛋白质已经与某个配体结合了,那么配体的原子会以HETATM行的形式出现在 PDB 文件中。其中我们在RDKIT工具进行pdb转json过程中,xxx_ligands.sdf文件中的多个配体位姿,会同pdb文件中的数据进行保存,本身pdb文件中不止包含蛋白质结构,大多文件都有相关配体信息,而sdf文件中的配体信息是不包含在内的,在推理过程中我们是希望配体和蛋白质的氨基酸链有一个结合,故而一并保存经数据预处理后送往推理模型中。
- 这个文件描述了一个蛋白质的三维结构,包含了蛋白质的氨基酸链和通过
命令解析:
bust 7ZTL_BCN_ligands.sdf -p 7ZTL_BCN_protein.pdb --outfmt long .评估配体与蛋白质的结合精度,通常会计算 RMSD 或其他指标来衡量配体与蛋白质的配对程度。下表是对应打印输出的具体信息与解释:
| 1. MOL_PRED loaded: | 表示配体分子预测数据已加载。这通常意味着程序已成功读取配体数据并开始分析。 |
| 2. MOL_COND loaded: | 表示配体的条件(如溶剂化、质子化等)已加载并进行预处理。 |
| MOL_TRUE loaded | 已加载真实分子 |
| 3. Sanitization: | 这一部分检查配体分子的标准化情况,确保分子没有化学或几何错误,例如非法的键合或不一致的原子类型。 |
| 4. InChI convertible: | 表示配体是否能转换为标准的InChI(国际化学标识符)。InChI是化学结构的标准化表示,确保分子结构能够以标准方式表示。 |
| 5. All atoms connected: | 表示所有原子都已连接,确保分子结构完整,没有断裂的键或孤立的原子。 |
| Molecular formula | 分子式计算 |
| Molecular bonds | 分子键计算 |
| Double bond stereochemistry | 双键立体化学 |
| Tetrahedral chirality | 四面体手性 |
| 6. Bond lengths: | 配体中各原子之间的键长信息。此字段确保键长符合常规化学规则,并检查是否存在不合理的键长。 |
| 7. Bond angles: | 配体中原子之间的键角信息,确保分子几何结构符合标准。 |
| 8. Internal steric clash: | 检查配体内部是否有空间冲突,即原子或原子组之间是否发生了重叠,违反了空间排斥原则 |
| 9. Aromatic ring flatness: | 检查配体中的芳香环是否平面化。芳香环通常应保持平面结构,如果不平面,可能会导致分子不稳定。 |
| 10. Double bond flatness: | 检查配体中双键是否平面化。双键的平面性对分子的稳定性至关重要。 |
| 11. Internal energy: | 配体的内部能量,通常是通过力场计算得出的,表示分子构象的稳定性。能量较高的构象通常是不稳定的。 |
| 12. Protein-ligand maximum distance: | 配体与蛋白质之间的最大距离。这帮助评估配体在蛋白质结合口袋中的位置,防止配体与蛋白质之间存在不合理的距离。 |
| 13. Minimum distance to protein: | 配体与蛋白质之间的最小距离。这个值对于确定配体是否可能与蛋白质发生相互作用至关重要。 |
| 14. Minimum distance to organic cofactors: | 配体与有机辅因子之间的最小距离。配体与辅因子之间的合理距离对其结合模式和稳定性很重要。 |
| 15. Minimum distance to inorganic cofactors: | 配体与无机辅因子之间的最小距离。 |
| 16. Minimum distance to waters: | 配体与水分子之间的最小距离。水分子可以在蛋白质-配体结合中扮演重要角色,合理的水分子分布对于稳定配体-蛋白质结合很重要。 |
| 17. Volume overlap with protein: | 配体与蛋白质之间的体积重叠。体积重叠较大可能表示配体和蛋白质之间存在空间冲突。 |
| 18. Volume overlap with organic cofactors: | 配体与有机辅因子之间的体积重叠。 |
| 19. Volume overlap with inorganic cofactors: | 配体与无机辅因子之间的体积重叠。 |
| 20. Volume overlap with waters: | 配体与水分子之间的体积重叠。 |
| RMSD ≤ 2Å | 均方根偏差(RMSD)小于等于 2Å (计算) |
1. bust: 这是执行分子对接或分析的工具命令。它可能是类似PoseBusters或其他分子对接 分析工具的命令行工具。
2. xxx_ligands.sdf: 这是输入的配体文件,使用SDF(Structure Data File)格式。 3. -p xxx_protein.pdb:这是指定输入蛋白质的PDB文件,xxx_protein.pdb是蛋白质文件, 包含目标蛋白质的三维结构。 4. --outfmt long: 这个选项指定了输出格式为long,即详细格式,输出配体与蛋白质之间的 各种相互作用信息以及配体的物理和化学属性。 总结:
这条命令的作用是通过bust工具,读取配体(.sdf文件)和蛋白质(.pdb 文件),然后根据--outfmt long选项输出配体与蛋白质之间的详细相互作用信息。输出包括配体的几何形状、能量、与蛋白质和辅因子的距离、体积重叠情况、是否存在内部冲突等信息。这些数据有助于进一步评估配体在蛋白质结合口袋中的物理合理性,并帮助识别潜在的结构问题。
sdf文件理解
以5S8I_2LY_ligand.sdf文件为例,其中包含了分子的结构坐标、元素类型、键连接信息等。SDF 文件用于存储分子的三维结构和相关的化学信息。以下是对这个文件的详细解释:
1. 文件头部信息
5S8I_2LY_A_1501
RDKit 3D- 5S8I_2LY_A_1501: 这个可能是配体或分子的标识符,通常是由蛋白质复合体的ID和配体编号组成的。
- RDKit 3D: 表示该分子是使用 RDKit 库生成的三维结构。
2. 分子坐标和元素信息
13 14 0 0 0 0 0 0 0 0999 V2000- 13 14: 13个原子和14个键。
- 0 0 0 0 0 0 0 0 0 0 0 0 0: 这些是保留的位置信息,通常与原子/键的特征有关。
- 0999 V2000: 文件格式的版本和识别码。
3. 原子坐标和元素类型
接下来的行列出了原子的三维坐标和元素类型。例如:
-24.0230 15.1580 30.4680 C 0 0 0 0 0 0 0 0 0 0 0 0
每行代表一个原子的详细信息:
- -24.0230 15.1580 30.4680: 该原子的x, y, z坐标。
- C: 元素符号,表示碳原子。
- 后面的数字表示该原子的其它特征,如原子类型、标记等。
4. 键连接信息
1 2 1 0
2 3 1 0
3 4 1 0
4 5 2 0
这些是键的连接信息:
- 每行代表一个化学键。
- 第一列和第二列表示连接的原子编号(例如,原子1和原子2连接)。
- 第三列表示键的类型(1表示单键,2表示双键等)。
- 第四列通常是用于额外信息的字段。
5. M END 和 $$$$
M END
$$$$- M END: 表示该分子描述的结束。
- $$$$: 用于表示SDF文件的结束标记。
BUST测试中的使用信息
BUST(Binding Uncertainty and Structure Test)是分子对接测试的一种方法,它用来验证分子在与目标蛋白结合时的可靠性。以下是BUST测试中可能使用的几个关键数据:
1. 原子坐标和元素类型:
- 分子坐标(例如 -24.0230 15.1580 30.4680)用于确定配体的三维位置,配体对接到蛋白质的过程中,原子的位置是必需的。BUST测试会检查这些坐标是否与实验数据(例如X射线晶体学数据)相符合,验证配体的结构和姿态。
2. 键连接信息:
- 键的连接信息(如 1 2 1 0)用于描述分子内部原子如何连接,这对于重建分子的几何结构至关重要。在BUST测试中,这些连接信息用于确保对接的配体结构没有发生不合实际的几何变化。
3. 键类型:
- 键的类型(如单键、双键)影响配体的刚性或柔性。在BUST测试中,这些信息用于模拟配体的自由度,验证其在对接中的灵活性。
分子姿态验证
BUST测试用于验证通过对接程序得到的配体姿态是否合理。具体来说,BUST会:
- 检查生成的分子姿态是否能够恢复配体在已知的蛋白质-配体晶体复合物中的真实位置。
- 验证分子姿态的几何稳定性,确保分子没有发生物理上不可能的形变(例如,过于扭曲的分子结构)。
- 确认分子与蛋白质的相互作用(如氢键、疏水相互作用等)是否合理。
通过对比预测的配体姿态与实验获得的配体-蛋白质复合物(例如通过X射线晶体学获得的结构),BUST测试帮助确认分子对接的准确性。
pdb文件理解
这段数据是一个以5SAK_ZRY_protein.pdb为例的PDB(Protein Data Bank)格式的蛋白质结构文件,PDB文件格式广泛用于描述蛋白质、核酸和其他生物大分子的三维结构。下面是对这些信息的详细解析:
PDB文件用于描述蛋白质等大分子的三维结构,它的格式是固定的,每一行通常对应一个原子(ATOM)或一个配体(HETATM)等信息。我们逐行解析每个部分的含义:
CRYST1行(晶体学信息)
CRYST1 45.330 73.690 52.740 90.00 109.70 90.00 P 1 21 1 0
- CRYST1: 该行提供晶体学的基本信息,通常用于描述晶体的单元格参数。
- 45.330, 73.690, 52.740: 晶胞的三条轴的长度 (单位为Ångström)。
- 90.00, 109.70, 90.00: 三个角度的度数(单位为度,α, β, γ)。
- P 1 21 1: 表示空间群信息(P1空间群,常用于非对称的晶体结构)。
- 0: 表示该数据的对称性信息。
ATOM/ HETATM行(原子或配体信息)
ATOM 行:
ATOM 1 N SER A 1 51.427 -12.547 61.084 1.00 15.78 A N 每一列的意义:
1. ATOM: 表示该行描述的是一个蛋白质的原子。如果是配体,它通常会显示为HETATM。
2. 1: 原子的序号(从1开始编号)。
3. N: 原子的元素符号,这里是氮 (Nitrogen)。
4. SER: 氨基酸的三字母代码,这里是丝氨酸(Serine)。
5. A: 链ID,表示这是蛋白质链的A链。如果蛋白质有多个链,会有不同的链ID(A, B, C...)。
6. 1: 氨基酸的序号,表示这是第一位氨基酸。
7. 51.427 -12.547 61.084: 原子的空间坐标(x, y, z)坐标,单位是Ångström。
8. 1.00: 表示该原子的占据度,1.00表示完全占据。
9. 15.78: B因子(也叫热振动因子,表示原子的位置不确定性,较大的B因子表示较大的位移)。
10. A: 表示原子所在的残基的插入位点。如果有插入位点,字母A、B、C等表示不同的插入位点。
11. N: 原子的元素类型(这里再次为氮)。
HETATM 行:
HETATM 4668 S DMS A 401 29.456 8.284 57.787 0.73 19.20 B S
- HETATM: 表示这个原子是一个配体或非标准氨基酸(如DMS,二甲基硫)。非标准氨基酸和配体通常使用HETATM行来表示。
- 4668: 配体原子的序号。
- S: 配体的元素符号(硫)。
- DMS: 配体的名称,这里是二甲基硫(DMS)。
- A: 链ID,表示该配体是蛋白质A链的一部分。
- 401: 配体的位置编号。
- 29.456 8.284 57.787: 配体原子的空间坐标(x, y, z)坐标。
- 0.73: 占据度。
- 19.20: B因子,表示原子在空间中的热振动程度。
- B: 插入位点。
- S: 该原子的元素类型。
TER 行:
标识蛋白质的链的结束。这意味着接下来没有其他同一链的原子。
CONECT 行:
连接原子的顺序,表示不同原子之间的共价键或接触。
END:
文件的结束标志
BUST 测试应用:
BUST(Basic Unit of Structure Testing)是一种用于验证蛋白质或大分子结构合理性的工具,它会通过分析PDB文件中的各种信息来进行不同的验证,主要验证结构是否符合物理化学规则。例如,它会检查:
1. 空间协调性:通过PDB文件中的原子坐标,BUST可以验证结构是否符合几何学要求,如原子之间的距离、角度、链之间的合理接触等。
2. B因子分析:通过原子的B因子(热振动因子)来评估模型的质量,BUST可以判断模型的热震荡和合理性。过高或过低的B因子可能表示数据有问题。
3. 键长和角度的合理性:BUST可以使用PDB中的原子信息来检查分子中所有键的长度和角度是否符合已知的化学规则。
4. 非标准氨基酸和配体的分析:通过PDB中的HETATM行,BUST可以检查配体或非标准氨基酸的结构是否正确以及是否与蛋白质的结构合理结合。
5. 蛋白质的二级结构:BUST可以基于PDB中的原子信息推断蛋白质的二级结构,如螺旋、β-折叠等,并检查其合理性。
BUST测试通过比较结构中的不同元素(如氨基酸残基、原子、配体)以及它们在空间中的分布情况,确保结构模型符合分子生物学的基本规则和物理化学性质。这是确保结构数据准确性的一个重要步骤。
RMSD(Root Mean Square Deviation)解析
1. RMSD的基本定义:
RMSD(Root Mean Square Deviation,均方根偏差)是衡量两个分子或结构之间差异的标准方法,特别是在分子对接、分子模拟和结构比较等领域。它量化了两个分子或结构在三维空间中的相似性,通常用于比较两组原子的位置。
RMSD的计算方法如下:
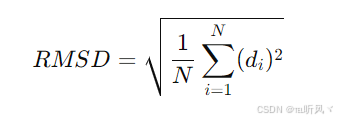
其中:
- N是原子对的数量(例如,两个分子中匹配的原子数)。
- 是对应的原子对之间的空间距离(即第i个原子对的偏差)。
- ![]() 表示对所有原子对偏差的平方和。
表示对所有原子对偏差的平方和。
- 最后,取平均值并开平方得到RMSD值。
2. RMSD的物理意义:
RMSD提供了两个分子结构之间在三维空间中的“平均”偏差,数值越小,表示两者越相似。它是通过计算每个对应原子对之间的距离差来获得的。
- 小的RMSD值:表示两个结构之间的差异较小,它们在空间上的重合度较高。通常认为,RMSD值小于 2 Å(埃)时,结构差异不大,意味着两个结构是非常相似的。
- 大的RMSD值:表示两个结构之间差异较大。较大的RMSD值通常表示两者在空间位置上有显著的不同。
3. RMSD的常见用途:
1. 分子对接结果的评估:
- 在药物设计和分子对接中,RMSD是评估对接结果是否合理的重要指标。如果一个小分子配体与目标蛋白的对接结构的RMSD值很低(通常小于2-3 Å),则表示对接结果比较准确,预测的配体结构与实验结构之间的差异很小。
- 例如:如果通过对接得到的配体姿势与已知的蛋白质-配体复合物的X射线晶体结构相比,RMSD值很小,那么这个对接结果就可以认为是可靠的。
2. 分子模拟中的结构比较:
- 在分子动力学模拟(MD)过程中,RMSD常常用于比较模拟过程中某一分子系统的当前结构与初始结构之间的差异。通过观察RMSD随时间的变化,可以了解分子是否发生了显著的构象变化。通常,RMSD稳定或增长缓慢说明分子趋于稳定,过大的RMSD可能意味着系统尚未达到平衡状态。
- 例如:一个分子系统的RMSD从开始的较高值(通常意味着分子结构从初始状态发生了较大的变化)逐渐下降并保持平稳,表示分子在模拟过程中逐步收敛到其稳定的构象。
3. 多构象比较:
- RMSD常用于比较多个分子构象,尤其是在筛选和优化候选化合物时。它能够帮助研究者判断不同构象之间的差异,选择最优的构象或理解分子如何从一个构象过渡到另一个构象。
- 例如:在药物筛选过程中,通过计算不同配体姿势的RMSD,可以评估它们在与目标蛋白结合时的相似性,筛选出结构相似、最稳定的候选分子。
4. 蛋白质结构预测与比对:
- 在蛋白质结构预测中,RMSD用于比较计算预测的结构与实验测定的结构之间的差异。结构预测的精度通常通过RMSD来衡量。如果预测的结构RMSD较小,表示预测较为准确;如果RMSD较大,表示预测存在较大的误差。 - 例如:通过比较通过同源建模预测的蛋白质三维结构与实验解析的结构,可以评估同源建模方法的效果。
5. 重叠分析:
- RMSD可以用于分析分子之间的重叠程度,尤其是在比较不同的构象或多个分子(例如,多个配体或多个蛋白质链)时,RMSD可以作为度量它们空间重叠度的一个标准。对于某些高通量筛选(HTS)或蛋白质-配体对接,RMSD可以帮助确定是否存在多个构象的空间重叠,或者是否发现潜在的构象变化。
4. RMSD的限制:
- 对比原子选择的依赖性:RMSD值取决于选取的原子对。例如,在比较蛋白质和配体时,可以选择仅比较配体的核心原子或整个结构的所有原子。不同的选择会导致不同的RMSD结果。
- 无法区分构象之间的细微差异:虽然RMSD值能够量化两个结构之间的差异,但它对结构的局部变化(例如,局部旋转或小幅度的变形)可能不敏感,尤其是在结构差异较小时。 - RMSD数值过高时的局限性:当RMSD值较高时,往往表明结构差异较大,但它并不总是能完全 描述分子间的相似性。有时候,仅凭RMSD并不足以全面反映结构的相似性,还需要结合其他分析方法(如能量、构象空间的分析等)。
5. RMSD的计算实例:
假设我们有两个配体结构,一个是预测结构,另一个是来自实验的参考结构。通过对比这两个结构的原子位置,计算RMSD,可以得出两者之间的平均偏差。 - 如果 RMSD = 0.5 Å,则表示这两个结构非常相似,差异极小。
- 如果 RMSD = 3 Å,则表示这两个结构在空间位置上有明显差异,可能代表不同的构象或对接结果。
总结:
RMSD是一个广泛应用于生物信息学、药物设计、分子建模等领域的重要指标,它帮助科学家们量化和评估分子或蛋白质之间的空间结构差异。通过计算两个结构之间的RMSD,可以了解它们在三维空间中的相似性或差异性,进而评估分子对接的准确性、分子模拟的稳定性以及蛋白质结构预测的精度。尽管RMSD是一个非常有用的工具,但它的局限性也要求我们在使用时需要谨慎,结合其他分析方法共同进行结构评估。


















 2009
2009

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









