



 本文详细介绍了使用MCScanX进行物种间共线性分析的过程,包括软件下载安装、文件准备、数据库构建、BLAST比对及MCScanX运行步骤,并提供了下游分析和绘图方法。
本文详细介绍了使用MCScanX进行物种间共线性分析的过程,包括软件下载安装、文件准备、数据库构建、BLAST比对及MCScanX运行步骤,并提供了下游分析和绘图方法。
一篇不知道拖了多久的笔记……欠下的债早晚还是要还的……
0.软件下载及安装
官网:
参考教程:
其实之前已经有两位前辈写过教程了的,我这只是再给需要的朋友举个例子打个样。详情请戳下面两个链接
下载软件:
解压:
unzip MCScanX.zip
cd到MCScanX后,按照
介绍的方法,给
msa.h
dissect_multiple_alignment.h
detect_collinear_tandem_arrays.h
这三个文件前面添加上#include <unistd.h>
即,用vim将文件打开后在开头加上上述的参数段。这个错误的原因是,MCScanX 不支持64位系统。
然后make一下就安装好了
1.文件准备
从TAIR上下载TAIR10的pep和gff3数据,要先用notepad++的正则替换功能(vim的批量替换如果会用的话也是可以的)将蛋白质的基因ID后面的注释信息给删掉,否则会影响后续的工作。接下来处理gff3文件。
先看一眼原始的数据长啥样:
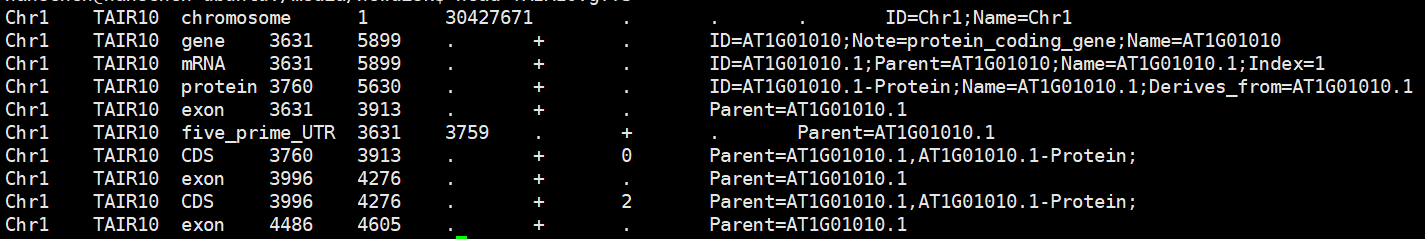
我们需要的是第1、4、5列和第9列的ID部分
用awk、grep和vim的全局替换功能可以得到所需要的简化版gff3文件:
cat TAIR10.gff3 |awk '/gene/ {print $1 "\t" $3 "\t" $4 "\t" $5 "\t" $9 }'> tmp.gff3
$3是用来标记用的,我们只取含有gene的列
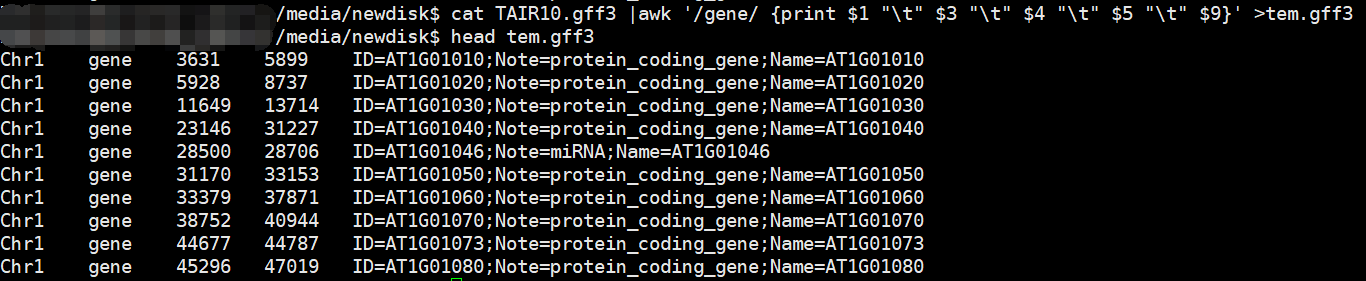
但是会有一些问题,比如:
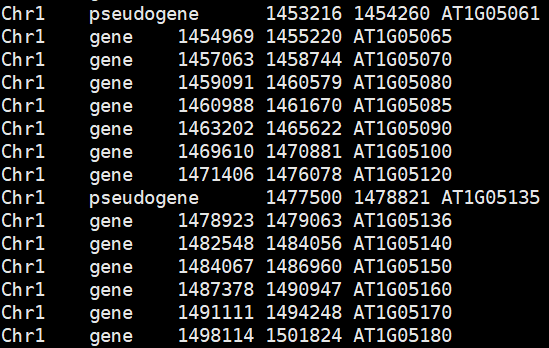
在搜索gene的时候只要包含gene的词就会被检索到,于是有以下三个混进来的:
pseudogene
transposable_element_gene
mRNA_TE_gene
于是我就:
grep -v 'mRNA_TE_gene' tmp2.gff3 > tmp3.gff3
grep -v 'pseudogene' tmp3.gff3 > tmp4.gff3
grep -v 'transposable_element_gene' tmp4.gff3 > tmp5.gff3
由于当时学艺不精比较菜就只能先暂时这样用一下了……

现在想想好像在上一步awk的时候可以:
cat TAIR10.gff3 |awk '/^gene/ {print $2 "\t" $0}' > tmp.gff3
加上一个“^”表示以gene开头,应该就可以免去上面那繁琐的几步了,最后生成的时候再倒一下回来就好了。
可以用
grep -v "^gene" tmp3.gff3
来检测是否还包含有非gene的行。
经过一系列的awk操作之后获得最终的结果:
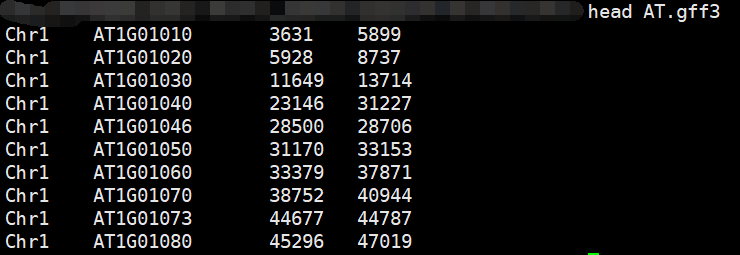
另一个物种如法炮制即可。
updates:也可以这么写,三步搞定:
awk '$3 == "gene" {print $1 "\t" $3 "\t" $4 "\t" $5 "\t" $9}' TAIR10.gff3 > new.gff3
用vim全局替换处理ID列的冗余信息
用awk将信息位置调换正确
2.建库
makeblastdb -in AT.pep -dbtype prot -parse_seqids -out ATdb
因为我需要做的是maca对拟南芥的共线性比较,所以只需要对拟南芥建库即可。(如果需要物种内也进行共线性比较的话这里需要把两个物种的蛋白质文件cat到一起来建库)
3.比对
nohup blastp -query maca.pep -db ATdb -out AT_maca.blast -evalue 1e-10 -num_threads 30 -outfmt 6 -num_alignments 5 &
参数稍微解释一下。
-query 被比对的物种的蛋白质文件
-db 上一步建的库
-out 输出结果
-evalue 设置输出结果的e-value值,值越小相似度越高。
-num_threads 比对所用的线程数
-outfmt 输出的格式
-num_alignments 输出比对上的序列的最大值条目数
这一步还是要点时间的。记得挂nohup。
4.运行MCScanX
将之前的两个物种的gff3文件cat成一个文件后命名为AT_maca.gff
将上一步生成的AT_maca.blast和AT_maca.gff放到新建的AT_maca文件夹内
MCScanX ./AT_maca
之后就会生成一个叫AT_maca.collinearity的文件和一个AT_maca.html文件夹。
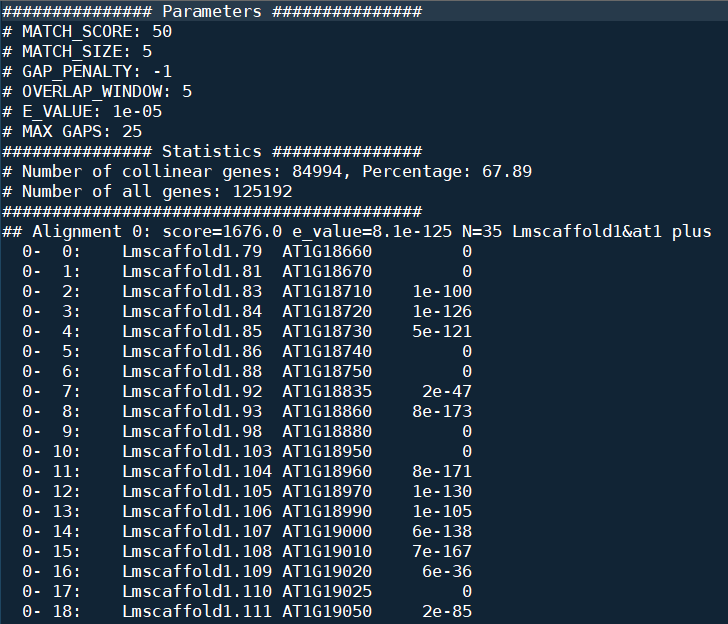
AT_maca.html里面是这样滴:

5.下游分析
java circle_plotter -g AT_maca.gff -s AT_maca.collinearity -c circle.ctl -o AT_maca_circle.png
这一步需要修改修改circle.ctl文件
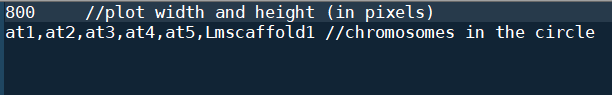
只要把你希望画出来的序列的ID给写进去就好了
可以画dot图,bar图和circle图,个人觉得circle图最好看了~
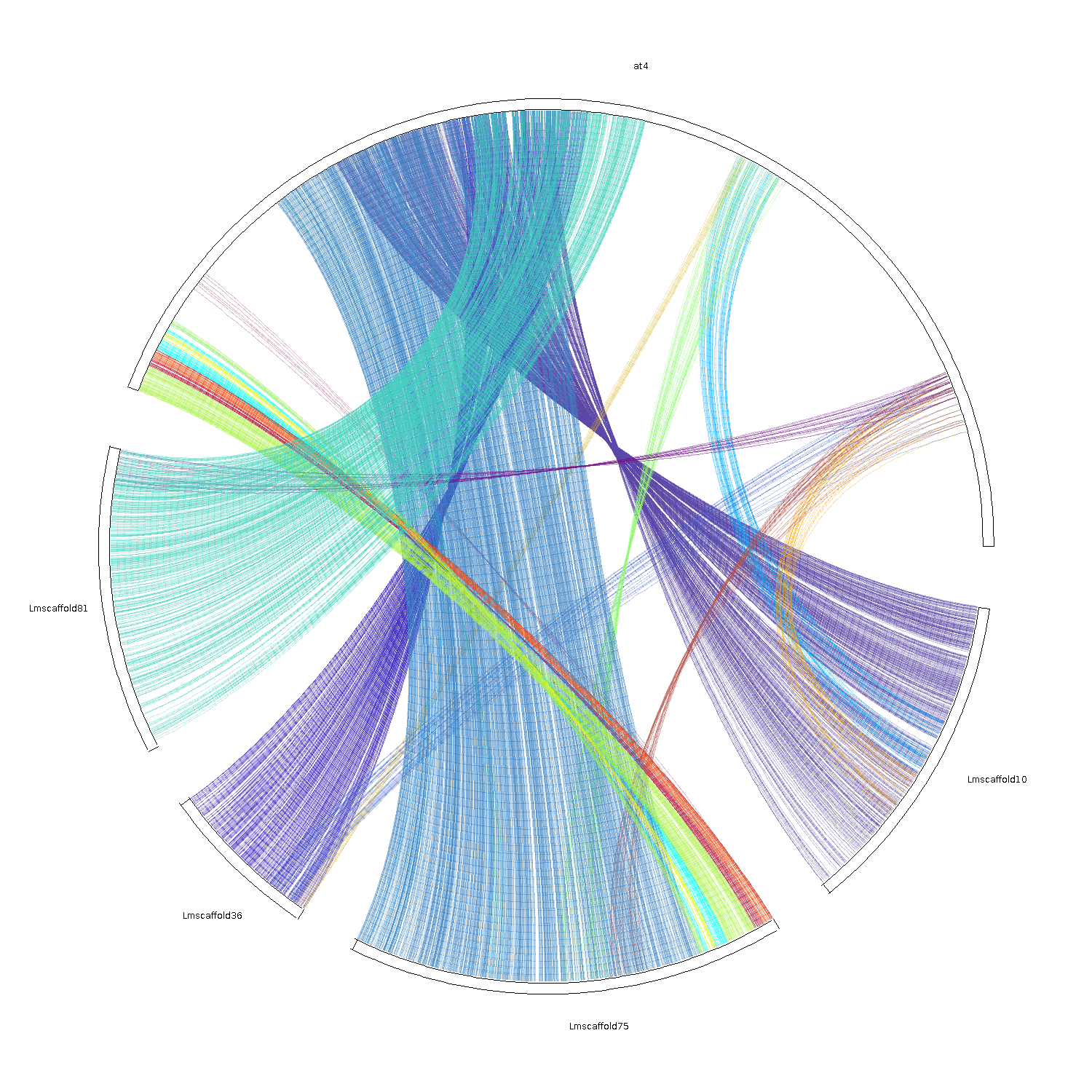
大概就是这样了。如果有什么错误的地方欢迎讨论指出哟。

















 5503
5503

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









