



 Samtools是一组用于操作SAM、BAM和CRAM格式的实用程序,包括文件转换、排序、合并和索引等功能。本文介绍了Samtools的安装、SAM与BAM文件、常用命令,如View、Sort、Merge、Index等,帮助用户更好地理解和使用Samtools进行数据分析。
Samtools是一组用于操作SAM、BAM和CRAM格式的实用程序,包括文件转换、排序、合并和索引等功能。本文介绍了Samtools的安装、SAM与BAM文件、常用命令,如View、Sort、Merge、Index等,帮助用户更好地理解和使用Samtools进行数据分析。
Samtools 是一组实用程序,用于操作 SAM(序列比对/映射)、BAM 和 CRAM 格式的比对。它在格式之间进行转换,进行排序、合并和索引,并且可以快速检索任何区域中的读取。被广泛应用在分析流程中。接下来小编就为大家介绍一下Samtools吧!
安装方法
方法一:
下载安装包一步步安装,链接地址为:https://github.com/samtools/samtools/releases/(在此就不做演示)
方法二:
使用conda(建议使用这种方法,简单方便)
conda install samtools
安装成功后,在终端输入samtools,出现如下界面后,表示安装成功.

SAM文件和BAM文件
1.SAM(Sequence Alignment/Map)格式是一种通用的比对格式,用来存储reads到参考序列的比对信息。SAM是一种序列比对格式标准,由sanger制定,是以TAB为分割符的文本格式。主要应用于测序序列mapping到基因组上的结果表示,当然也可以表示任意的多重比对结果。SAM分为两部分,注释信息(header section)和比对结果部分(alignment section)。
2.BAM是SAM的二进制文件,bam文件优点:bam文件为二进制文件,占用的磁盘空间比sam文本文件小;利用bam二进制文件的运算速度快。
对比如下:

查看bam文件头部:
samtools view -h test.bam | head

HD:VN表示版本,SO表示排序方式。
SQ:SN表示参考序列的名称,LN表示参考序列的长度
PG:比对时使用的工具指令。(这里小编用的是hiast2)
常用命令
1.View
view命令的主要功能是:将sam文件转换成bam文件;然后对bam文件进行各种操作,比如数据的排序(不属于本命令的功能)和提取(这些操作 是对bam文件进行的,因而当输入为sam文件的时候,不能进行该操作);最后将排序或提取得到的数据输出为bam或sam(默认的)格式。view命令中,对sam文件头部的输入(-t或-T)和输出(-h)是单独的一些参数来控制的。将sam文件转为bam文件(没有header的sam文件不能转换成bam文件)
基本使用方法:
samtools view -S test.sam -b > test.bam

将bam文件转为sam文件
基本用法:
samtools view -h -o test.sam test.bam
-b 默认下输出是 SAM 格式文件,该参数设置输出 BAM 格式-S 默认下输入是 BAM 文件,若是输入是 SAM 文件,则最好加该参数,否则有时候会报错。-o 输出文件名
提取比对到参考序列上的比对结果F
基本用法:
samtools view -bF 4 test.bam >test.F.bam

-F 数字4代表该序列没有比对到参考序列上 数字8代表该序列的mate序列没有比对到参考序列上
比对到反向互补链的reads
基本用法:
samtools view -f 16 test.bam|head -1

比对到正向链的reads
基本用法:
samtools view -F 16 test.bam|head -1

统计共有多少条reads(pair-end-reads这里算一条)参与了比对参考基因组
基本用法:
samtools view -c test.bam

筛选出比对失败的reads,看序列特征
基本用法:
samtools view -f 4 test.bam|cut -f10 |head -3

筛选出比对质量值大于30的情况
基本用法:
samtools view -q 30 test.bam |awk '{print $1,$5}'|head -3
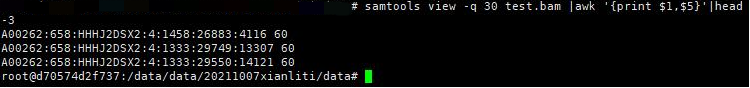
筛选出比对成功,但是并不是完全匹配的序列
基本用法:
samtools view -F 4 test.bam |awk '{print $6}'|grep '[IDNSHPX]'|head -5

2.Sort
对bam文件进行排序
基本用法:
samtools sort -@ 10 -o test.bam test.sam
-@ 设置排序和压缩是的线程数量,默认是单线程。-o 输出文件名

3.Merge
当有多个样本的bam文件时,可以使用samtools的merge命令将这些bam文件进行合并为一个bam文件。Merge命令将多个已经排序后的bam文件合并成为一个排序的且保持所有输入记录并保持现有排序顺序的bam文件。
基本用法:
samtools merge out.bam test1.bam test2.bam test3.bam
4.Index
为了能够快速访问bam文件,可以为已经基于坐标排序后bam或者cram的文件创建索引,生成以.bai或者.crai为后缀的索引文件。必须使用排序后的文件,否则可能会报错。另外,不能对sam文件使用此命令。如果想对sam文件建立索引,那么可以使用tabix命令创建。
基本用法:
samtools index -b test.bam
-b 创建bai索引文件,未指定输出格式时,此参数为默认参数


5.Faidx
对fasta文件建立索引,生成的索引文件以.fai后缀结尾。该命令也能依据索引文件快速提取fasta文件中的某一条(子)序列.如果没有指定区域,faidx命令就创建文件索引并生成后缀为.fai的索引文件。如果指定区域,那么就是生产并显示fasta格式的子序列。输入文件可以使BGZF压缩格式的文件。输入文件中的序列要有不同的名称。如果不是这样,即存在相同名称的序列,在建立索引的过程中将发出有关重复序列的警告而且生产的同名子序列的信息都要被第一个同名子序列的信息覆盖。
基本用法:
samtools faidx ref.fasta


生成了索引文件fasta.fai,是一个文本文件,分成了5列。

第一列是序列名称
第二列为序列长度
第三列是OFFSET,第一个碱基的偏移量,以0为起始值。序列所在的位置”,因为该数字从上往下逐渐变大,最后的数字是genome.fasta文件的大小
第四列LINEBASES,除了最后一行以外,其他代表序列的碱基数,单位为bp;
第五列行宽(LINEWIDTH),除了最后一行以外,其他代表序列的长度,包括换行符。
6.Tview
tview能直观的显示出reads比对基因组的情况,和基因组浏览器有点类似。
基本用法:
samtools tview test.bam ref.fasta
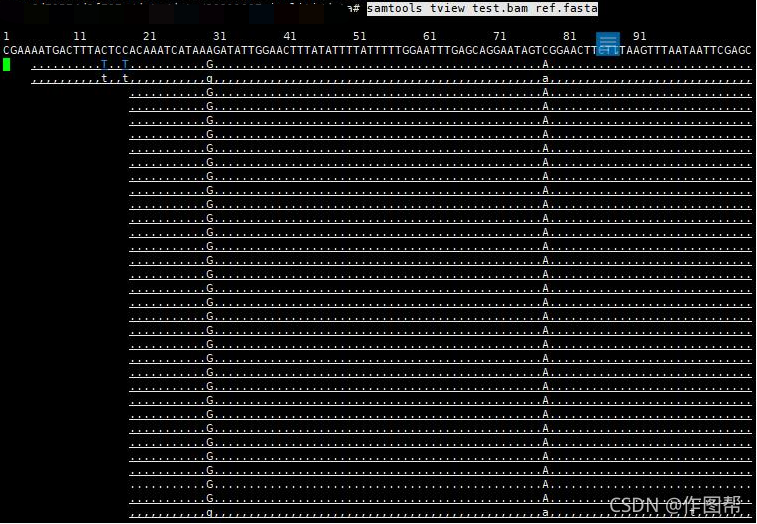
当给出参考基因组的时候,会在第一排显示参考基因组的序列,按下 g ,则提示输入要到达基因组的某一个位点。Ctrl+H 向左移动1kb碱基距离;Ctrl+L 向右移动1kb碱基距离。可以用颜色标注比对质量,碱基质量,核苷酸等。30~40的碱基质量或比对质量使用白色表示;20~30黄色;10~20绿色;0~10蓝色。使用点号‘.‘切换显示碱基和点号;使用r切换显示read name等如果没有参考序列则第一排都是用N表示

7.Flagstat
统计输入文件的相关数据并将这些数据输出至屏幕显示。每一项统计数据都由两部分组成,分别是QC pass和QC failed,表示通过QC的reads数据量和未通过QC的reads数量。以“PASS + FAILED”格式显示。
基本用法:
samtools flagstat test.bam

QC pass的reads的数量为68039736,未通过QC的reads数量为零,意味着总reads数为68039736
Mapped:比对到参考基因组上的reads数量
Paired:双端reads数据数量
Read1:R1的数量
Read2:R2的数量
Properly:完美匹配的reads数
PE中两条都比对到参考序列上的reads数单独一条匹配到参考序列上的reads数,和上一个相加,则是总的匹配上的reads数PE中两条分别比对到两条不同的参考序列的reads数PE中两条分别比对到两条不同的参考序列的reads数(比对质量>=5)
8.Depth
得到每个碱基位点或者区域的测序深度,并输出到标准输出。depth命令计算每一个位点的测序深度并在标准显示设备中显示。注意:使用此命令之前必须先samtools index。
基本用法:
samtools depth test.bam > result.depth.txt
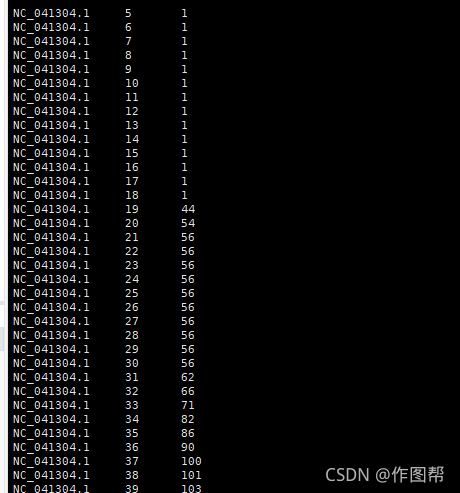
第一列为序列名称
第二列为位点
第三列为覆盖深度
9.Mpileup
该命令用于生成bcf文件,再使用bcftools进行SNP和Indel的分析。
基本用法:
samtools mpileup -C 50 -f ref.fasta test.bam

第一列:参考序列名称
第二列:位置
第三列:参考碱基
第四列:比对上的reads数
第五列:比对情况
第六列:比对上的碱基质量
第五列的解释:
.代表与参考序列正链匹配
,代表与参考序列负链匹配
ATCGN代表在正链上不匹配
Atcgn代表在负链上不匹配
*代表模糊碱基
代表匹配的碱基是一个read的开始;""后面紧跟的ascii码减去33代表比对质量;这两个符号修饰的是后面的碱基,其后紧跟的碱基(.,ATCGatcgNn)代表该read的第一个碱基。
$代表一个read的结束,该符号修饰的是其前面的碱基。
正则式+[0-9]+[ACGTNacgtn]+代表在该位点后插入的碱基,比如上例中在scaffold_1的2847后插入了9个长度的碱基acggtgaag。表明此处极可能是indel
正则式-[0-9]+[ACGTNacgtn]+代表在该位点后缺失的碱基
10.Fastq
将bam文件转为fastq格式
基本用法:
samtools fastq test.bam > test.fastq

以上就是关于Samtools的介绍啦,更详细的用法及说明请参考官方文档:http://samtools.sourceforge.net/samtools.shtml,有问题可以联系图图哦~


















 2814
2814

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









