




 本文介绍了TUTUCLOUD网站提供的在线染色体标记工具,用于基因定位分析。用户可以通过简单的步骤上传.txt或.csv文件,设置图片尺寸,并下载PDF格式的矢量图进行后期编辑。该平台适用于遗传学研究中的染色体标记。
本文介绍了TUTUCLOUD网站提供的在线染色体标记工具,用于基因定位分析。用户可以通过简单的步骤上传.txt或.csv文件,设置图片尺寸,并下载PDF格式的矢量图进行后期编辑。该平台适用于遗传学研究中的染色体标记。
染色体标记
基因定位是遗传学研究中的重要环节,染色体的标记是基因定位分析中重要的组成部分。TUTUCLOUD网站提供在线标记染色体的工具,可直观地表达某个或多个染色体中所含的基因名称以及基因所在的位置。
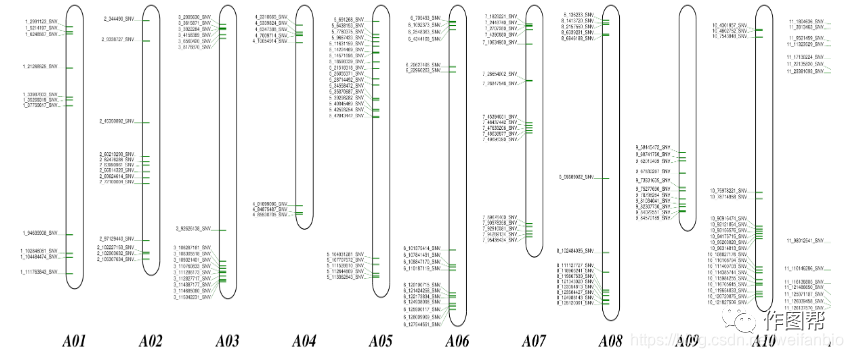
TUTU云工具使用
小编和他的小伙伴们开发了一个在线的作图小网站——云图图(www.cloudtutu.com,免费的哦~),操作步骤如下:
①登录网址:www.cloudtutu.com(推荐使用360或者谷歌浏览器)
②输入用户名和密码(小编已经为大家填好了,如果不显示可添加文末二维码添加小编获取),输入验证码后即可登录;
③登录后在工具一栏里找到染色体标记,点击进入;
④请按照界面右侧的说明书或者下文进行操作。
Step 1 上传文件
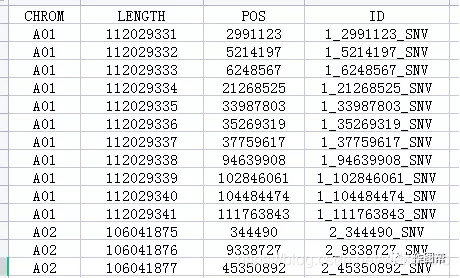
※目前平台仅支持.txt(制表符分隔)文本文件或者.csv文件的文件上传。(平台可对不规范的数据格式进行部分处理,但还是请您尽量按照示例数据的格式调整数据,以便机器可以识别)
a)准备一个数据矩阵(形式参照示例数据);
b)表格需要带表头和列名,每一行为染色体名称,每一列为各项指标名,如基因ID、SNV、长度、位置等等;
c)请提交txt(制表符分隔)文本文件或者.csv文件。操作方法为:全选excel中的所有内容(ctrl+A),复制到记事本中,将记事本文件另存后点击“上传”按钮上传该文件。
Step 2 设置参数
2.1 图片宽度:按需求自行设置
2.2 图片高度:按需求自行设置
 最低0.47元/天 解锁文章
最低0.47元/天 解锁文章


















 2420
2420

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









