



供稿 | 理想换柴米
审稿 | 吉星
目录
-
ATOMICA 模型学习生物分子交互的通用表征,揭示疾病机制并预测蛋白质功能,推动药物发现和暗蛋白组研究。
-
DrugAgent 利用多智能体和大型语言模型,实现了可解释且高精度的药物靶点相互作用预测。
-
FFADW 融合序列和网络特征,提高了蛋白质相互作用预测的准确性。
-
Genomic Tokenizer (GT) 利用密码子进行分词,提升了 Transformer 模型在 DNA 序列分析中的效率和生物学解释性。
-
知识蒸馏技术有效压缩分子性质预测模型,在保持甚至提升预测精度的同时,显著降低模型大小和训练时间。

1. ATOMICA:跨模态生物分子交互的通用表征学习模型
ATOMICA 是一种几何深度学习模型,它学习原子级表征,统一了蛋白质、核酸、小分子、离子和脂质等多种生物分子交互的框架。该模型利用超过 200 万个交互复合物进行自监督去噪训练,学习原子、化学块和界面层级的分层嵌入,并将其泛化到不同分子模态。
ATOMICA 生成特定模态的交互组网络 (ATOMICANET),基于交互界面的相似性连接蛋白质。这些网络揭示了共享的疾病通路,并有助于预测疾病相关蛋白。例如,它在基于脂质的网络中检测到与哮喘相关的钠通道,在基于离子的网络中检测到与白血病相关的锌指结构。对于自身免疫性疾病,如多发性硬化症和周围神经病变,ATOMICANET 以 100% 的命中率识别了高置信度靶点,例如电压门控钾通道和钠通道。
ATOMICA 的潜在空间根据物理原理组织原子和化学特征,无需监督即可恢复与元素周期表、氨基酸化学和核酸特性一致的模式。ATOMICA 支持对交互进行组合推理,类似于词嵌入中的向量运算,即使蛋白质序列不同,也能捕获复合物之间的语义相似性。使用 ATOMICASCORE 指标,该模型可以零样本识别对分子间相互作用贡献最大的残基,在结合位点识别方面优于大型蛋白质语言模型(如 ESM-2)。跨多种分子模态的预训练显著提高了泛化能力,例如,ATOMICA 将蛋白质-DNA 结合位点恢复的 AUPRC 从 0.24 提高到 0.71。
此外,ATOMICA 通过预测超过 2,600 个配体结合位点(包括新的 C4 锌指基序和血红素结合跨膜细胞色素)来注释暗蛋白组(以前未表征的蛋白质区域)。这些暗蛋白注释涵盖了 1,200 多个物种,并利用结构而非序列恢复了进化上古老的功能。
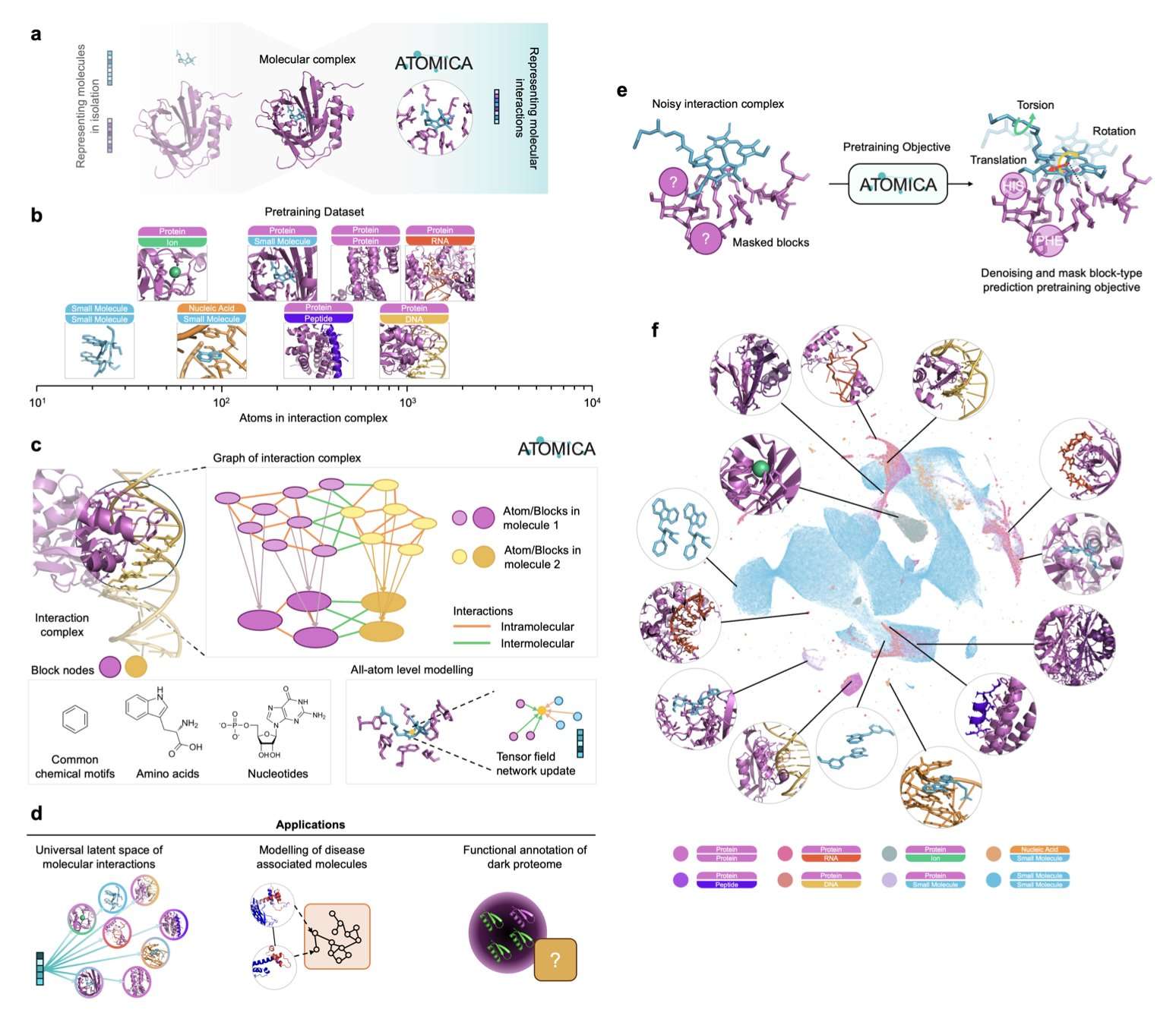
ATOMICA 的局限性包括依赖高质量的结构数据以及对柔性、无序蛋白质区域的覆盖有限。未来的工作旨在整合非结构交互数据和基于序列的特征,以扩展其适用性。ATOMICA 为交互分子的生成建模和界面引导的治疗设计奠定了基础,它将分子尺度的几何结构与疾病和蛋白质功能的系统级理解联系起来。
💻Code: https://github.com/mims-harvard/ATOMICA
📜Paper: https://www.biorxiv.org/content/10.1101/2025.04.02.646906v1
2. DrugAgent:基于多智能体的药物靶点相互作用预测
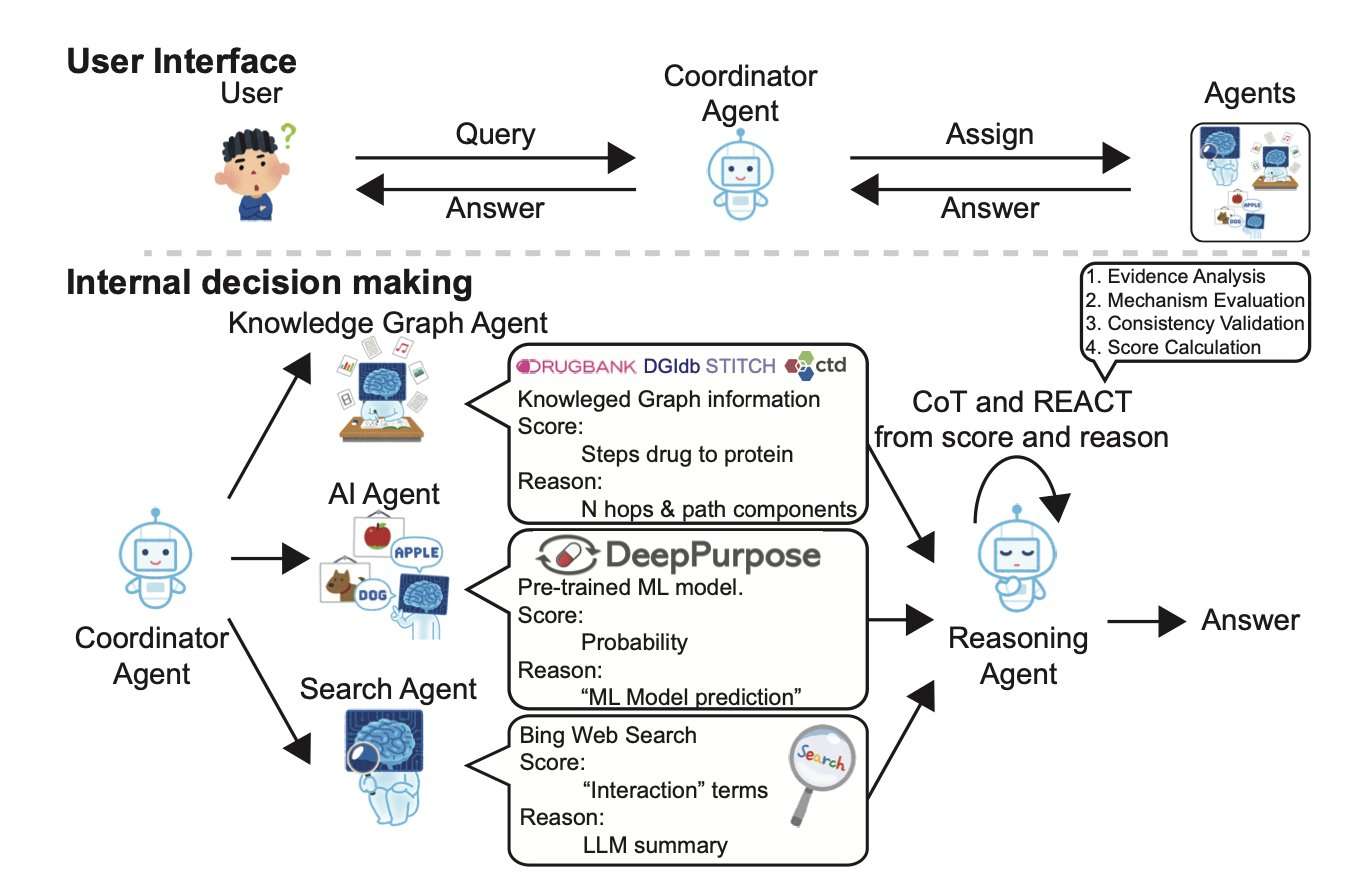
DrugAgent 是一个创新的多智能体系统,旨在预测药物靶点相互作用(DTI)。研究者整合了机器学习(ML)、知识图谱和文献证据等多重视角,并利用思维链(Chain-of-Thought)和 ReAct 框架实现了可解释推理。不同于黑盒模型,DrugAgent 对每个预测都输出人类可读的解释,使其适用于临床决策和监管环境等对透明度要求高的场景。
该系统由五个专门的智能体组成:协调器、AI(基于 ML)、KG(知识图谱)、搜索(文献)和推理。每个智能体都贡献一个分数及其基本原理,这些信息最终被整合为 DTI 预测结果。在激酶 - 化合物基准测试中,DrugAgent 的 F1 分数(0.514)比非推理 GPT-4o mini 基线高出 45% (0.355),且特异性高达 0.978,这对于最大限度地减少药物发现中的假阳性至关重要。
AI 智能体使用 DeepPurpose 和消息传递神经网络(MPNN)以及 CNN,根据 SMILES 字符串和蛋白质序列预测 DTI 概率,并在 BindingDB 上进行二元结合亲和力分类训练。KG 智能体则从 DrugBank、CTD、STITCH 和 DGIdb 构建统一图谱,计算基于路径的相互作用分数,强调药物和靶点节点之间生物学相关的路径。搜索智能体使用基于关键字的相关性启发式方法和基于 GPT 的摘要,从 Bing 检索和评分证据,提供真实世界的背景和潜在的临床支持。推理智能体使用 LLM 推理链整合所有证据,计算加权分数,验证一致性,并提供最终决策。例如,即使 ML 置信度接近于零,它也可以根据图谱和文献支持预测中等强度的相互作用。
消融研究表明 DrugAgent 具有鲁棒性:移除 AI 智能体会导致准确性急剧下降,而 KG 和搜索智能体则对减少假阳性和提高精度有显著贡献。除了优越的指标外,DrugAgent 还模拟了协作研究团队,每个智能体都充当专家。这种模块化设计便于扩展,例如添加检索增强生成(RAG)智能体或患者特定数据输入。尽管 token 成本比简单模型高 10 倍,但 DrugAgent 的可解释性和均衡的指标使其在现实世界的生物医学应用中更有价值,尤其是在高风险决策依赖于理解模型基本原理的情况下。该框架灵活、可复现且可扩展,使用 AutoGen、DeepPurpose、RDKit 和多个 LLM 在消费级 Mac 设备上实现。所有智能体输出都遵循严格的模板,以确保系统完整性。
💻Code: https://anonymous.4open.science/r/DrugAgent-B2EA
📜Paper: [2408.13378] DrugAgent: Multi-Agent Large Language Model-Based Reasoning for Drug-Target Interaction Prediction
3. FFADW:精准预测蛋白质相互作用的新方法
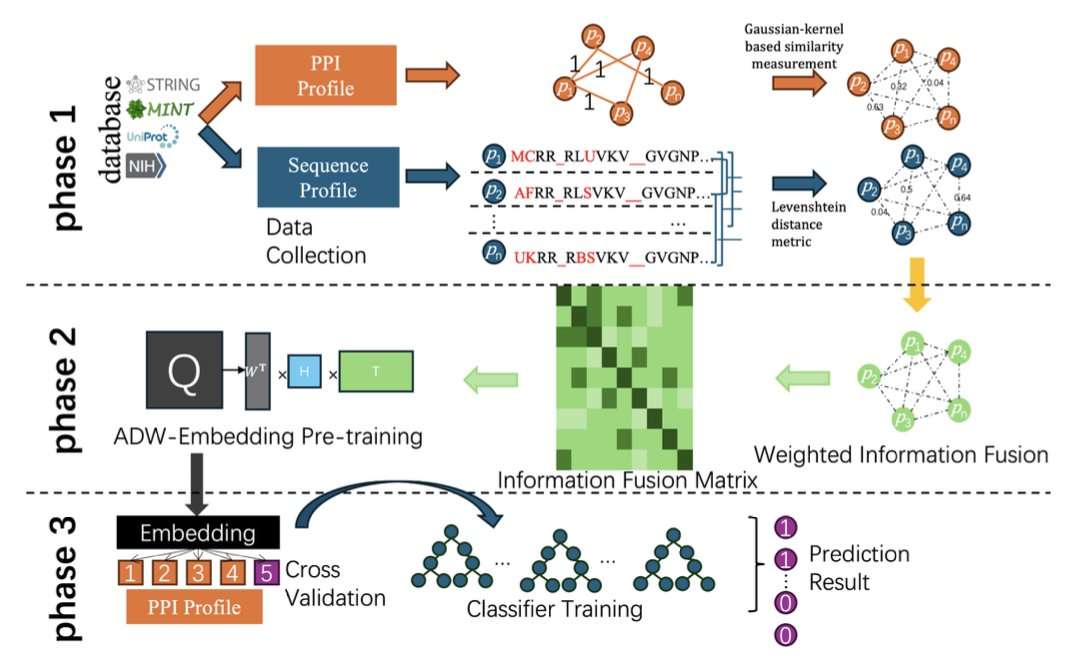
FFADW 是一种用于蛋白质相互作用 (PPI) 预测的新方法,它融合了蛋白质序列相似性和网络拓扑信息。作者利用 Levenshtein 距离提取序列特征,并通过高斯核计算网络特征,然后通过可调节的加权策略将这两种互补的生物数据类型组合成统一的、降噪的表示。该加权融合由可调参数 α 控制,允许在序列和网络贡献之间进行动态平衡,从而提高了模型在不同数据集上的灵活性和泛化能力。
Attributed DeepWalk 用于学习整合节点属性和网络结构的低维嵌入,增强了表示质量,且无需依赖深度学习,保持了方法的轻量级和可解释性。研究者在三个基准数据集(S. cerevisiae、Human 和 H. pylori)上评估了 FFADW 的性能,并使用 XGBoost 作为分类器,分别实现了 95.56%、98.68% 和 88.2% 的高准确率。尤其在 Human 数据集上,AUC 达到 0.994,接近完美。
与 Bio2Vec、GcForest-PPI 和 EResCNN 等现有先进方法相比,FFADW 在几乎所有关键指标(准确率、MCC、AUC)上都表现更优。大量的实验表明,特征融合持续改进了样本聚类和分类性能,t-SNE 可视化和 ROC 曲线验证了这一点。此外,FFADW 在多种分类器(SVM、RF、NB、XGB)上都表现稳健,统计分析证实 XGBoost 在大多数数据集上具有优越且具有统计学意义的性能。
FFADW 兼顾了可解释性和预测能力,非常适合计算资源有限或数据集太小而无法进行深度学习的研究人员。作者建议未来的改进可以包括自动 α 优化以及与注意力机制或 GNN 的集成,以处理更大规模的生物网络。
📜Paper: Feature fusion with attributed deepwalk for protein–protein interaction prediction | Scientific Reports
💻Code: https://github.com/StacyMYCao/FFADW
4. 基于生物学原理的 DNA 序列分词器:Genomic Tokenizer
Genomic Tokenizer (GT) 提出了一种基于生物学原理的 DNA 序列分词方法,其核心思想是使用密码子(三个核苷酸序列)作为分词单元,这与分子生物学的中心法则相符。与传统的字符或 k-mer 分词器不同,GT 能够识别起始和终止密码子,并将同义密码子分配给相同的标记,同时将内含子和框外区域视为 UNK 标记。这种策略在减少词汇量的同时,保留了生物学相关性。
GT 在 HuggingFace tokenizer 框架内实现,可以无缝集成到现有的基于 Transformer 的 DNA 分析流程中,并支持诸如掩码语言建模和序列分类等任务。该分词器支持自定义起始/终止密码子及内含子处理方式,使其能够适应不同的生物体,包括原核生物和线粒体基因组。
在使用肺癌相关变异数据集进行的分类实验中,与字符分词相比,GT 对长序列长度表现出更强的鲁棒性,并在较长序列任务中表现更优。虽然字节对编码 (BPE) 实现了最高的整体性能,但其庞大的词汇量带来了高昂的计算成本。GT 在生物学洞察力、计算效率和紧凑的词汇量之间取得了平衡。
GT 分词避免了重叠 k-mer 分词器中常见的掩码语言建模中的冗余和信息泄漏问题,从而获得更清晰的训练信号和潜在的更好的泛化能力。GT 的生物学基础使其能够更好地模拟框移突变、同义替换和终止增益变异——这些都是从遗传数据预测表型影响的关键特征。初步比较突出了 GT 在生物建模方面的优势,并表明与纯数据驱动的分词器相比,GT 在基因组学任务的基础模型训练中具有潜在优势。
GT 是开源的,可以通过 PyPI 安装,并鼓励在基因组数据集和 Transformer 架构(包括 HyenaDNA 等长上下文模型)中进行更广泛的探索。
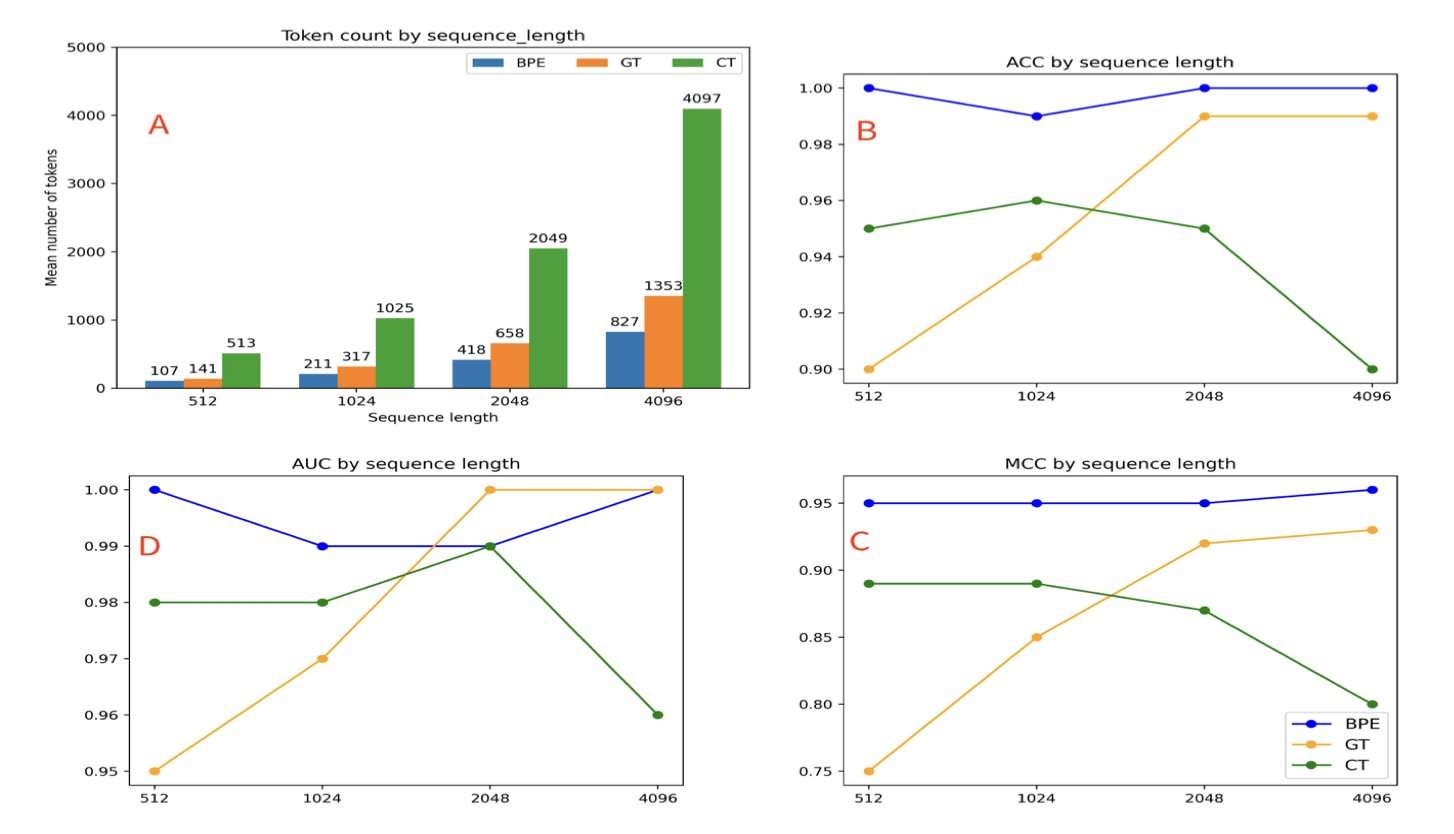
📜Paper: https://www.biorxiv.org/content/10.1101/2025.04.02.646836v1
💻Code: https://github.com/dermatologist/genomic-tokenizer

5. 知识蒸馏提升分子性质预测:模型更小,精度更高
研究者提出了一种将知识蒸馏(KD)应用于分子性质预测的综合框架,在回归任务中 R² 值提升高达 70%,同时减小了模型大小和训练时间。该方法使精简的学生模型(最高可缩小 2 倍)在量子数据集(QM9)和实验基准(ESOL、FreeSolv)上达到或超过复杂教师模型的性能,证明了其在特定领域和跨领域设置中的有效性。
该框架在 SchNet、DimeNet++ 和 TensorNet 三种 GNN 架构上均一致地提升了嵌入对齐和预测精度,尤其对于原子化能和溶解度等复杂性质的预测。在 QM9 特定领域设置中,DimeNet++ 对于较简单的量子性质,使用较小的学生模型时 R² 增益高达 90%,而 SchNet 在使用较大的学生模型预测复杂性质时增益最大。
从 QM9 训练的教师模型到实验数据集(如 ESOL(logS)和 FreeSolv(ΔGhyd))的跨领域迁移表明,KD 有助于弥合理论数据和经验数据之间的分布差距,SchNet 在溶解度预测上实现了约 65% 的 R² 增益。嵌入分析显示,KD 下学生模型和教师模型嵌入之间的余弦相似度显著提高,证实了跨模型和数据集的成功知识迁移和结构表示学习。
KD 尤其增强了对非 QM9 类分子的泛化能力,即 QM9 中不存在的原子或基团,这表明其对类药物或化学多样性化合物的实用性。该框架使用结合了 MAE 和余弦相似度损失以及可学习不确定性权重的损失函数,确保了任务自适应优化,无需手动调整。
与剪枝或迁移学习相比,KD 提供了一种结构化且稳健的模型压缩方法,在保留关键化学知识的同时实现更快的推理,这对于高通量筛选至关重要。这项工作突出了 KD 在分子回归中的可扩展性、与现代架构的兼容性,以及其在未来少样本和基于 Transformer 的分子建模应用中的潜在作用。
📜Paper: https://advanced.onlinelibrary.wiley.com/doi/10.1002/advs.202503271
💻Code: https://github.com/PEESEgroup/Knowledge-Distillation-For-Molecular-Properties
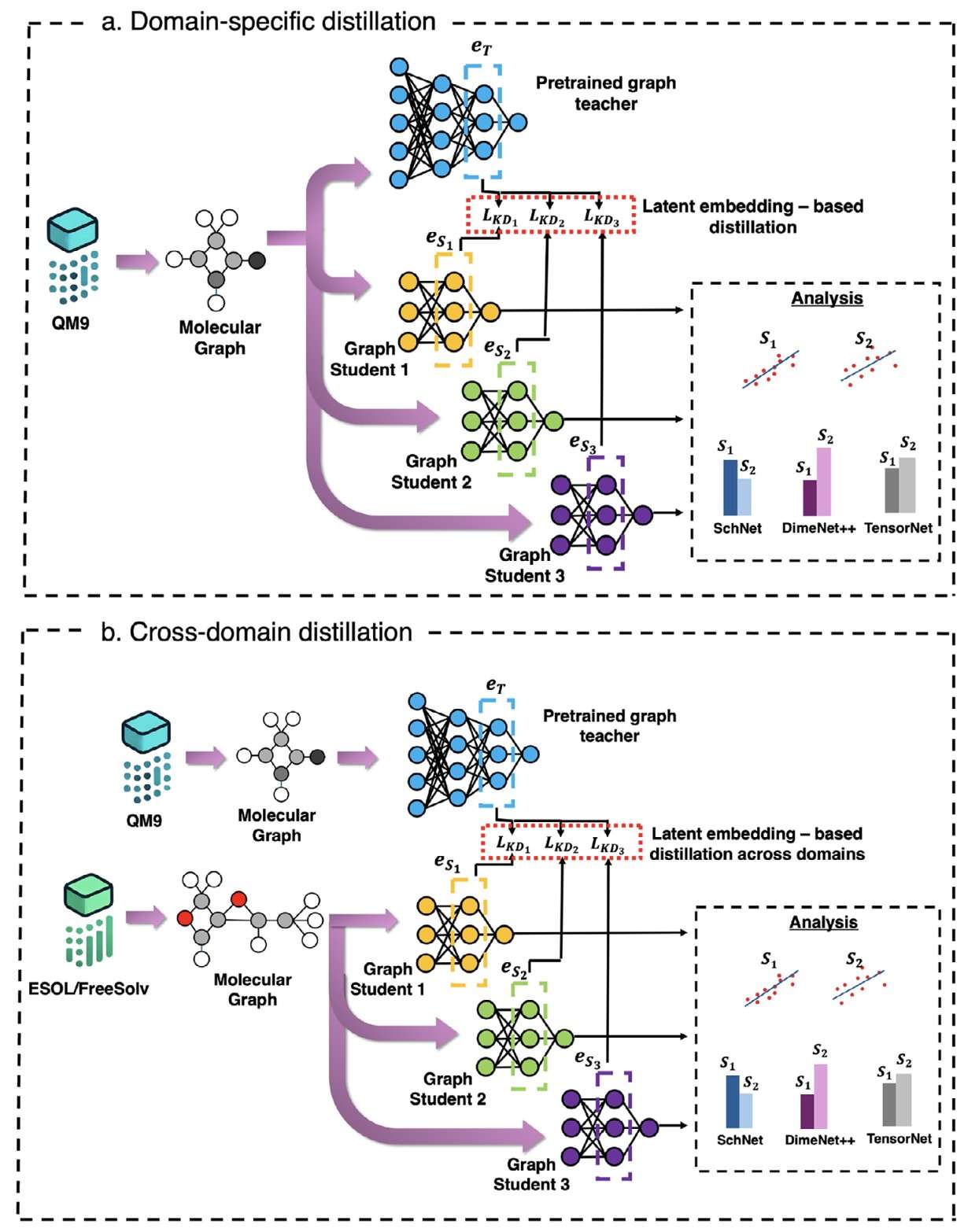
— 完 —

















 1240
1240

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









