



模块一:基础理论与设计思想 (3小时)
学习目标: 理解现代蛋白质设计的理论基石,建立从“功能”到“结构”再到“序列”的正确设计观。
- 蛋白质折叠问题与经典设计:
- 回顾“CASP历史”以及蛋白质折叠问题的复杂性 。
- 介绍David Baker实验室与IPD的设计哲学:以最终功能为导向,设计自然界不存在的全新蛋白质结构 。
- 讲解Rosetta软件的核心思想,理解其作为蛋白质设计基石的历史地位与方法论

模块二:核心AI模型原理与代码实现 (6小时)
学习目标: 从原理和代码层面,彻底理解驱动现代蛋白质预测与设计的两大核心神经网络架构。
- 基础工具与环境:
- 掌握Numpy进行多维数组(Tensor)操作 。
- 学习PyTorch的核心概念:张量、自动求导机制
- 讲解服务器GPU与CUDA的基本工作原理,为后续的本地化部署打下基础。
- 关键模型架构拆解:
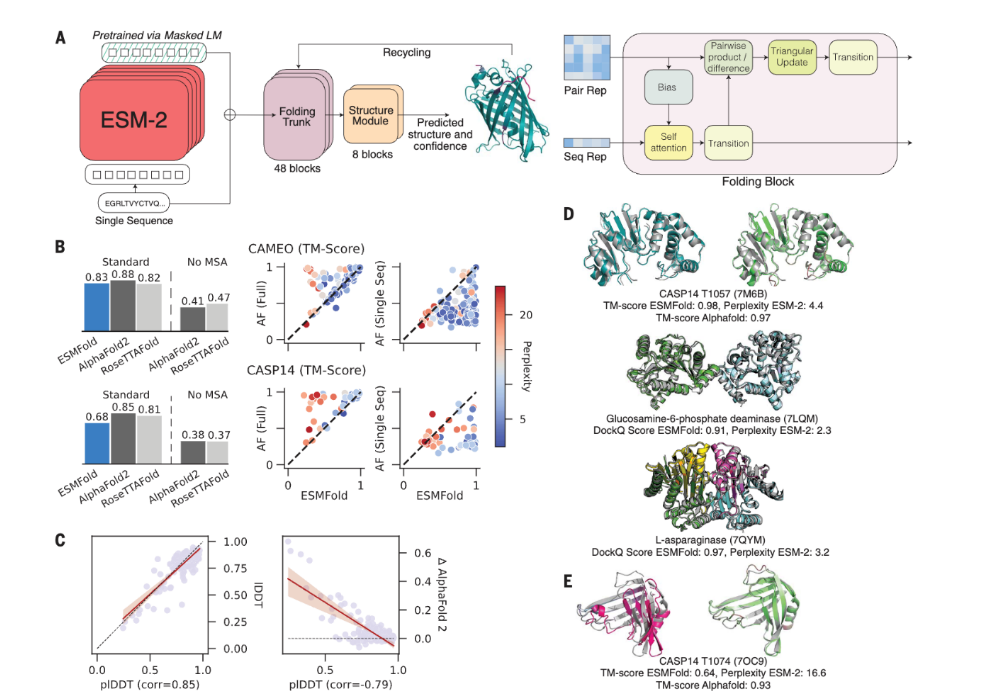
- Transformer与Attention机制:深入讲解Transformer模型如何通过自注意力机制捕捉序列中的长距离依赖关系,并分析其如何被AlphaFold 2用于整合多序列比对(MSA)信息和空间几何信息,构成Evoformer模块的核心 。
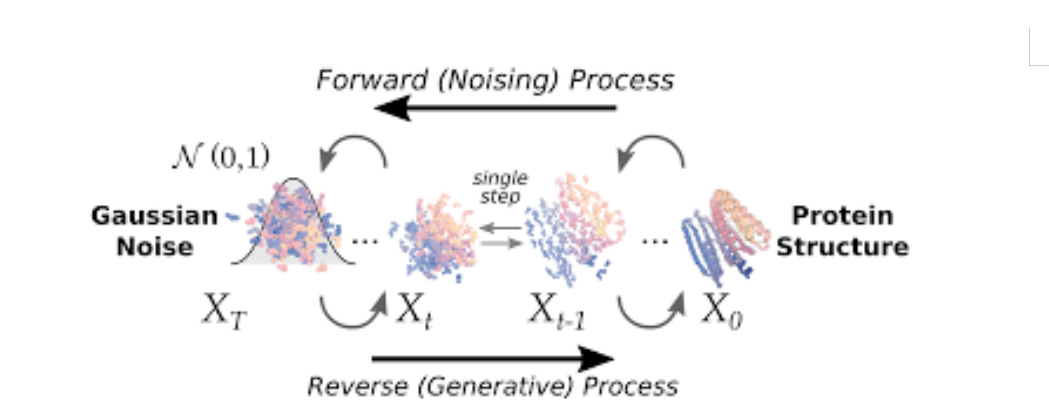
- 扩散模型 (Diffusion Model):详细阐述扩散模型的前向加噪(Forward Process)与反向去噪(Reverse Process)的数学原理 。重点讲解该模型如何被应用于RFdiffusion,实现从高斯噪声中逐步生成结构合理的蛋白质骨架

- 代码实践:所有模型原理都将配合Jupyter Notebook进行代码实操,确保学员不仅理解理论,更能动手实现。
模块三:前沿设计工具链:原理、部署与应用 (6小时)
学习目标: 掌握当前最高效的蛋白质设计“三要素”工具链,并完成本地化部署,具备独立开展计算设计的能力。
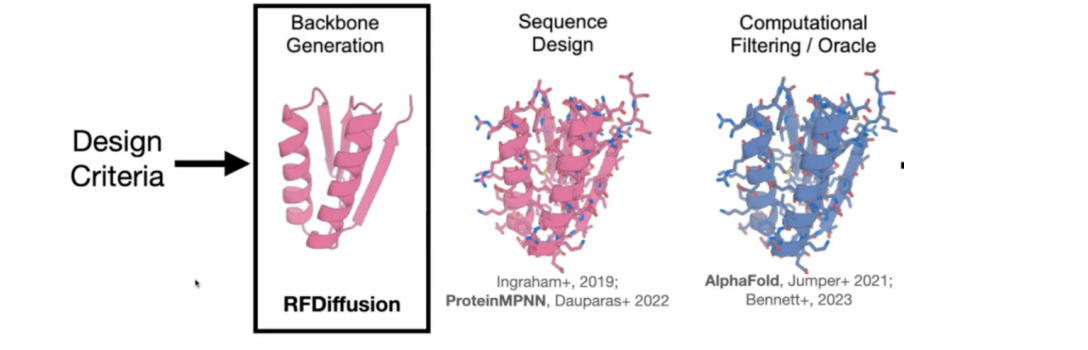
A. 结构生成 (RFdiffusion):
功能详解:学习RFdiffusion如何从随机噪声中生成全新的蛋白质骨架 。
实操技术:重点讲解几种核心设计模式的应用场景与参数设置:
无条件生成 (Unconditional generation):用于创造全新的拓扑结构。
基于骨架的生成 (Scaffolding):在已有的结构骨架上进行延申或构建。
功能位点限定生成 (Inpainting):在固定关键功能位点(如活性中心、结合界面)的前提下,生成包裹该位点的全新结构 。
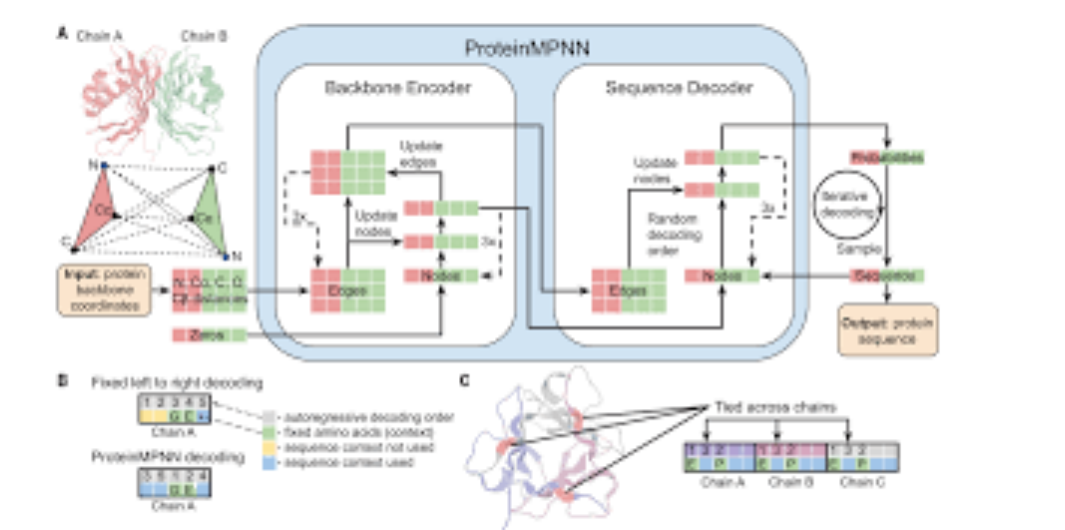
- B. 序列设计 (ProteinMPNN):
- 核心问题:讲解“蛋白质反向折叠 (Inverse Folding)”问题的挑战性,即如何为给定的骨架设计出能正确折叠的氨基酸序列 。
- 协同工作流:演示如何将RFdiffusion生成的骨架(Backbone)作为输入,利用ProteinMPNN设计出高稳定性的氨基酸序列,形成完整的“结构-序列”设计流程 。
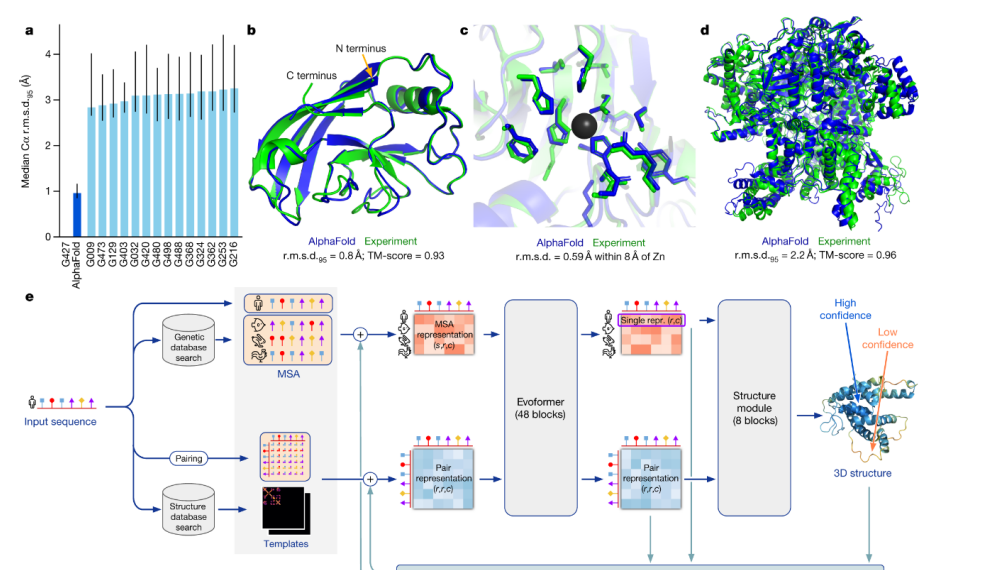
- C. 结构验证与功能拓展 (AlphaFold 3):
- 精度验证:学习使用AlphaFold 2/3验证由ProteinMPNN设计的序列,能否高精度地折叠回RFdiffusion所设计的初始结构 。重点讲解pLDDT和PAE图的解读,用于评估预测结果的置信度 。
- 功能预测:介绍AlphaFold 3的核心突破——其预测能力已从单一蛋白质扩展到蛋白质、DNA、RNA、配体等多种分子的复合物 。探讨如何利用这一特性,初步评估设计的蛋白质与靶点分子的相互作用,为药物设计提供关键信息。
模块四:应用方向专题实战 (15小时)
学习目标: 将前面学习的工具链应用到三个主流的生物医药研发场景中,解决具体问题。
- 专题一:从头设计功能性Binder (5小时)
- 技术流程:一个完整的综合性项目。学员将选择一个具体靶点,独立运用“RFdiffusion → ProteinMPNN → AlphaFold”的全套技术流程,从零开始设计一个能够特异性结合该靶点的全新小分子蛋白(Binder),完成一个标准化的de novo设计流程。
- 技术流程:一个完整的综合性项目。学员将选择一个具体靶点,独立运用“RFdiffusion → ProteinMPNN → AlphaFold”的全套技术流程,从零开始设计一个能够特异性结合该靶点的全新小分子蛋白(Binder),完成一个标准化的de novo设计流程。
- 专题二:酶的计算改造与性能优化 (5小时)
- 技术流程:学习利用同源结构数据库进行搜索,结合计算工具进行定向突变,以优化酶的催化活性、底物特异性或热稳定性。课程将介绍定向进化与适应性景观等核心概念 。
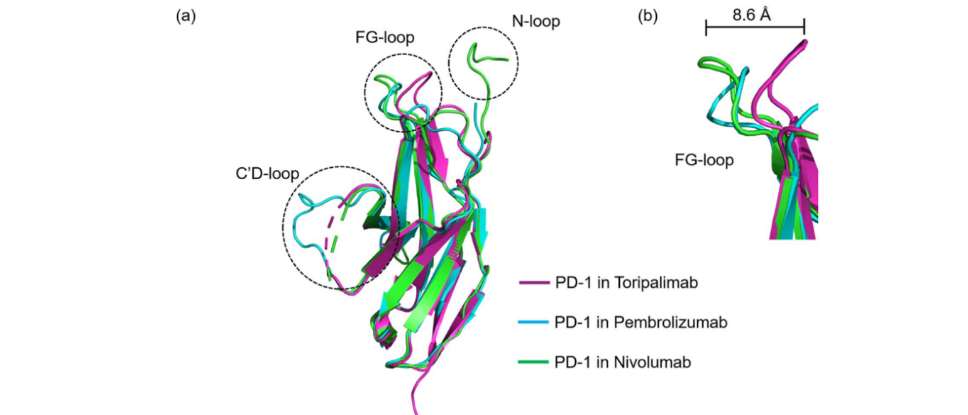
- 专题三:抗体药物的计算设计 (5小时)
- 技术流程:掌握计算辅助的抗体设计方法,包括靶点抗原的分析、互补决定区(CDR)的设计与优化,以及利用AlphaFold-Multimer等工具预测抗体-抗原复合物结构,评估亲和力与特异性 。
模块四:应用方向专题实战 (15小时)
总学习目标:通过三个独立的、项目驱动的专题,让学员完整地、端到端地执行计算蛋白质设计的流程。在课程结束时,学员不仅掌握了工具的使用,更能理解在不同应用场景下(酶、抗体、从头设计)的设计策略、关键考量和技术组合。
专题一:从头设计功能性Binder (5小时)
项目目标:执行一个完整的、工业界标准的de novo设计流程,针对一个选定的蛋白靶点,从零开始设计一个能够特异性结合其表面的全新小分子蛋白(Binder)。
核心概念:De novo设计、蛋白质-蛋白质相互作用(PPI)、形状互补性、计算流程的端到端整合。
技术流程详解:
- 靶点分析与结合位点选择:
- 选择一个感兴趣的靶点蛋白(如疾病相关蛋白)。
- 分析其表面性质,选择一个适合作为结合靶点的区域(通常是相对平坦、具有一定疏水性的表面)。
- 基于靶点的骨架生成 (RFdiffusion):
- 此为设计的核心创造步骤。学员将学习使用RFdiffusion的功能位点限定生成 (Inpainting) 模式 。
- 将靶点蛋白的结构作为固定的“环境”,RFdiffusion会在此环境的约束下,从噪声中“生长”出一个与靶点表面形状高度互补的全新蛋白质骨架。
- 序列填充与优化 (ProteinMPNN):
- 将上一步生成的最优骨架输入ProteinMPNN。
- ProteinMPNN会为这个全新的骨架设计出能够稳定折叠的氨基酸序列,完成从“形状”到“化学本质”的转化 。
- 双重计算验证 (AlphaFold):
- 步骤一:单体验证。将ProteinMPNN设计的序列输入AlphaFold,预测其单体结构。验证该序列能否独立折叠成我们设计的形状(高pLDDT得分,且RMSD与设计模型低)。
- 步骤二:复合物验证。使用AlphaFold-Multimer 预测设计的Binder和靶点蛋白的复合物。验证Binder能否如预期般结合到靶点的正确位置(低界面PAE得分)。
专题二:抗体药物的计算设计 (5小时)
项目目标:掌握计算辅助的抗体亲和力成熟(Affinity Maturation)流程,针对一个已知的抗体-抗原复合物,设计新的互补决定区(CDR)序列,以增强其结合能力。
核心概念:抗体结构域、CDR与框架区(FR)、抗原表位(Epitope)与互补位(Paratope)、抗体人源化、亲和力与特异性。
技术流程详解:
- 抗体-抗原复合物的结构分析:
- 从PDB中选取一个抗体-抗原复合物结构作为起始点。
- 利用可视化软件,精确识别构成结合界面的CDR loop,特别是贡献最大的CDR-H3。
- 详细分析界面上的氢键、疏水作用、盐桥等关键相互作用,理解亲和力的结构基础。
- CDR区域的序列设计:
- 固定抗体的框架区和抗原结构,仅针对CDR区域进行序列设计。
- 学员将使用ProteinMPNN等工具,在保持CDR loop骨架构象的同时,重新设计其氨基酸序列,以期发现能与抗原形成更优相互作用的新序列。
- 利用AlphaFold-Multimer进行复合物结构预测与评估:
- 将新设计的CDR序列整合回抗体,形成一个完整的突变抗体序列。
- 使用AlphaFold-Multimer 预测新设计的抗体与抗原的复合物结构。这是流程中最关键的一步。
- 学员将学习如何解读AlphaFold-Multimer的输出结果,特别是利用PAE(Predicted Aligned Error)图来评估界面预测的置信度,判断新设计的抗体是否仍能以正确的模式结合抗原。
- 亲和力排序与候选筛选:
- 通过比较不同设计方案的界面结合面积、预测的氢键网络、以及界面PAE得分等指标,对设计的抗体进行排序。
- 筛选出1-3个预测结合能力最强、且结构最可靠的候选抗体序列。
专题三:酶的计算改造与性能优化 (5小时)
项目目标:针对一个给定的酶,通过计算方法,设计出具有更高热稳定性或催化活性的突变体,并提供一套可供实验验证的候选序列。
核心概念:适应性景观 (Fitness Landscape) ,定向进化 ,酶催化与过渡态理论 (Transition State Theory) ,计算诱变 (In-silico Mutagenesis),折叠自由能 (ΔG)。
技术流程详解:
- 模板选择与结构准备:
- 从蛋白质结构数据库(PDB)中选取一个具有已知结构和功能的酶作为改造模板。
- 若无实验结构,将学习利用UniProt 寻找同源序列,并使用SWISS-MODEL 或本地AlphaFold进行结构预测,获得可靠的起始模型。
- 使用PyMOL 或ChimeraX 分析酶的活性位点、底物结合口袋以及潜在的不稳定区域(如高柔性loop)。
- 计算突变扫描与稳定性预测:
- 学习使用Rosetta等工具包中的能量函数,对酶的特定区域(或全长)进行计算饱和突变扫描。
- 核心技术点是计算每个突变体相对于野生型的折叠自由能变化 (ΔG)。学员将学习如何解读ΔG值,以筛选出能够提升蛋白质热稳定性的有益突变。
- 催化性能的计算评估:
- 对于活性位点附近的突变,将引入分子对接(Molecular Docking)方法,模拟底物或过渡态类似物与突变酶的结合情况。
- 通过比较结合能、关键相互作用(氢键、盐桥等)的变化,来半定量地预测突变对催化活性(kcat/KM)的潜在影响。
- 候选突变体的筛选与组合:
- 基于上述计算结果,学员将学习如何制定筛选策略,例如,优先选择同时提升稳定性且不损害(或提升)活性的突变。
- 探讨如何组合多个有益的单点突变,以获得性能提升更显著的多点突变体。

















 975
975

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









