






聚类分析和相关分析是两种常用的统计方法,它们在数据科学和医学研究中具有重要意义。热图是一种流行的可视化手段,用于展示这两种分析的结果。
首先,我们来了解一下热图。热图是一种以颜色的变化来表示数据矩阵或数据集中程度的图表工具。颜色越深表示数据集中度越高,颜色越浅表示数据集中度越小。热图常用于展示基因表达、蛋白质相互作用、代谢途径活性等方面的数据信息。
聚类分析的热图详解
聚类分析是一种将数据集中的对象分组的统计方法,目的是使组内对象的相似度尽可能高,而组间对象的相似度尽可能低。聚类分析的结果通常通过热图来展示,热图中的颜色深浅表示聚类成员之间的相似度高低或距离远近。下面通过一个示例来了解一下热图的制作流程,R代码如下:
#加载需要的R包
library(ggh4x)
library(ggplot2)
library(tidyverse)
library(ggdendro)
#准备数据,热图的输入是一个数值型矩阵
test = matrix(rnorm(200), 20, 10) #创建一个随机数矩阵
test[1:10, seq(1, 10, 2)] = test[1:10, seq(1, 10, 2)] + 3
test[11:20, seq(2, 10, 2)] = test[11:20, seq(2, 10, 2)] + 2
test[15:20, seq(2, 10, 2)] = test[15:20, seq(2, 10, 2)] + 4
colnames(test) = paste("Test", 1:10, sep = "") #命名列名Test1——Test10
rownames(test) = paste("Gene", 1:20, sep = "") #命名行名Gene1——Gene20
yclust <- hclust(dist(test)) #计算基于矩阵test行的距离关系进行的层次聚类的结果
xclust <- hclust(dist(t(test))) #计算基于矩阵test列的距离关系进行的层次聚类的结果
p = test %>%
#转换矩阵为数据框
as.data.frame() %>%
#将行名添加为一列
rownames_to_column() %>%
#将数据从宽格式转换为长格式,其中每一行包含一个样本和对应的基因表达值
pivot_longer(cols = 2:ncol(.),
names_to = "sample",
values_to = "exp") %>%
#以下开始使用ggplot2进行绘图
#首先ggplot()函数初始化一个绘图对象,aes()指定x轴和y轴的变量
ggplot(aes(x = sample,y = rowname))+
#geom_tile()函数绘制瓷砖图,fill = exp指定根据基因表达值来填充颜色
geom_tile(aes(fill = exp))+
#设定填充颜色的渐变,包括中点颜色、低值颜色和高值颜色
scale_fill_gradient2(midpoint = 2.5,
low = '#2fa1dd',
mid="white",
high = '#f87669') +
#将基于聚类结果yclust绘制的行的树形图添加到热图中
scale_y_dendrogram(hclust = yclust) +
#将基于聚类结果xclust绘制的列的树形图添加到热图中
scale_x_dendrogram(hclust = xclust,position = 'top') +
#设定图形的主题,包括隐藏网格线、更改背景颜色等
theme(panel.grid = element_blank(), #隐藏网格线
axis.line = element_blank(), #隐藏坐标轴线
axis.ticks = element_blank(), #隐藏坐标轴刻度线
axis.title = element_blank(), #隐藏坐标轴标题
panel.background = element_rect(fill = NA), #去除面板的背景颜色
legend.background = element_rect(fill = NA), #去除图例的背景颜色
plot.background = element_rect(fill = NA),) #去除绘图区域的背景颜色代码的运行结果如图所示。我们使用ggplot2完成了热图的绘制。这段代码生成的热图展示了基因表达数据中样本间和基因间的关系,并结合聚类树形图展示了基于样本和基因之间相似性的聚类结构。
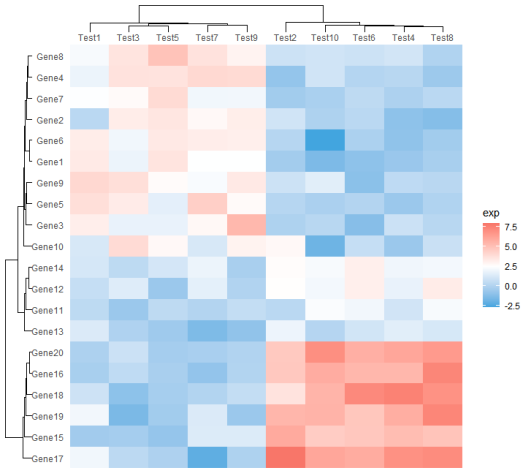
通过这个热图,可以直观地观察到不同基因在不同样本中的表达模式、相似性和聚类关系,有助于发现潜在的模式、规律和群组结构。同时,颜色渐变和聚类树形图也提供了额外的信息,有助于研究人员更好地理解和解释数据中的特点。
相关分析的热图详解
多个特征值之间的相关性关系可以通过相关性热图(correlation heatmap)有效地表征。相关性热图通过颜色的深浅来表示特征间相关系数的大小,常用于探索数据集中不同变量间的关系。它不仅能帮助我们直观地识别变量之间的显著正相关或负相关,还能帮助我们检测潜在的多重共线性问题。接下来,我们将使用ggplot2包绘制热图,以展示两个基因的相关性。该图展示了所有变量间的相关系数,颜色深浅表示相关系数的大小,并将显著的相关系数数值标记在图中。R代码如下:
#生成一个随机数矩阵作为示例数据
set.seed(123)
data <- matrix(rnorm(100), 10, 10)
#计算相关性矩阵
correlation_matrix <- cor(data)
#将相关性矩阵转换为长数据格式
cor_data <- as.data.frame(as.table(correlation_matrix))
colnames(cor_data) <- c("gene1", "gene2", "correlation")
#设置显著性水平
significance_level <- 0.5
#绘制相关性热图并标记显著的相关系数
ggplot(cor_data, aes(x = gene1, y = gene2)) +
#根据correlation填充色块的颜色
geom_tile(aes(fill = correlation)) +
#在热图的色块上添加筛选出相关性较为显著的文本
geom_text(data = subset(cor_data, abs(correlation) > significance_level),
aes(label = round(correlation, 2)), color = "black", size = 3) +
#定义色块的渐变颜色
scale_fill_gradient2(low = "#2fa1dd", mid = "white", high = "#f87669", midpoint = 0) +
#应用一个简洁的主题样式
theme_minimal() +
theme(axis.text.x = element_text(angle = 45, hjust = 1, size = 8, color = "black"), #指定x轴上文本标签的样式
axis.text.y = element_text(size = 8, color = "black"), #指定y轴上文本标签的样式
panel.grid = element_blank(), #移除面板背后的网格线
strip.text = element_text(size = 10)) + #设置面板分隔文本的字体大小
#添加图形的标题和坐标轴的标签
labs(title = "Correlation Heatmap", x = "Gene 1", y = "Gene 2") +
#将x轴的刻度标签放置在顶部
scale_x_discrete(position = "top") +
#固定坐标轴的比例
coord_fixed() +
#调整图例的样式
theme(legend.title = element_text(size = 10, face = "bold"),
legend.text = element_text(size = 8))代码的运行结果如图所示。
通过图,我们可以直观地观察到不同基因在不同样本中的表达模式、相似性和聚类关系,有助于发现潜在的模式、规律和群组结构。同时,颜色渐变和聚类树形图也提供了额外的信息展示,帮助研究人员更好地理解和解释数据中的特点。
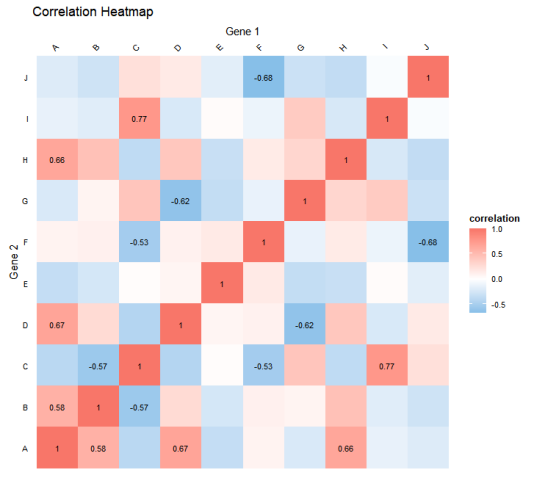
另外,多个特征值之间的相关性关系可以通过热图很好地进行表征。热图通过颜色的深浅来表示特征间相关系数大小的,它常用于探索数据集中不同变量间的关系。热图不仅能帮助我们直观地识别哪些变量之间存在显著的正相关或负相关,还能帮助我们检测潜在的多重共线性问题。接下来我们使用ggplot2包绘制热图展示两个基因的相关性的示例。展示所有变量间的相关系数,颜色表示相关系数大小,并将显著的相关系数数值标记在图中。
#生成一个随机数矩阵作为示例数据
set.seed(123)
data <- matrix(rnorm(100), 10, 10)
#计算相关性矩阵
correlation_matrix <- cor(data)
#将相关性矩阵转换为长数据格式
cor_data <- as.data.frame(as.table(correlation_matrix))
colnames(cor_data) <- c("gene1", "gene2", "correlation")
#设置显著性水平
significance_level <- 0.5
#绘制相关性热图并标记显著的相关系数
ggplot(cor_data, aes(x = gene1, y = gene2)) +
#根据correlation填充色块的颜色
geom_tile(aes(fill = correlation)) +
#在热图的色块上添加筛选出相关性较为显著的文本
geom_text(data = subset(cor_data, abs(correlation) > significance_level),
aes(label = round(correlation, 2)), color = "black", size = 3) +
#定义色块的渐变颜色
scale_fill_gradient2(low = "#2fa1dd", mid = "white", high = "#f87669", midpoint = 0) +
#应用一个简洁的主题样式
theme_minimal() +
theme(axis.text.x = element_text(angle = 45, hjust = 1, size = 8, color = "black"), #指定x轴上文本标签的样式
axis.text.y = element_text(size = 8, color = "black"), #指定y轴上文本标签的样式
panel.grid = element_blank(), #移除面板背后的网格线
strip.text = element_text(size = 10)) + #设置面板分隔文本的字体大小
#添加图形的标题和坐标轴的标签
labs(title = "Correlation Heatmap", x = "Gene 1", y = "Gene 2") +
#将x轴的刻度标签放置在顶部
scale_x_discrete(position = "top") +
#固定坐标轴的比例
coord_fixed() +
#调整图例的样式
theme(legend.title = element_text(size = 10, face = "bold"),
legend.text = element_text(size = 8))代码运行结果,如图所示,我们可以直观地了解基因之间的相关性情况,颜色变化展示了基因之间的关联性,帮助我们更好地理解数据中的模式和关系,为进一步的数据分析和探索提供了参考。
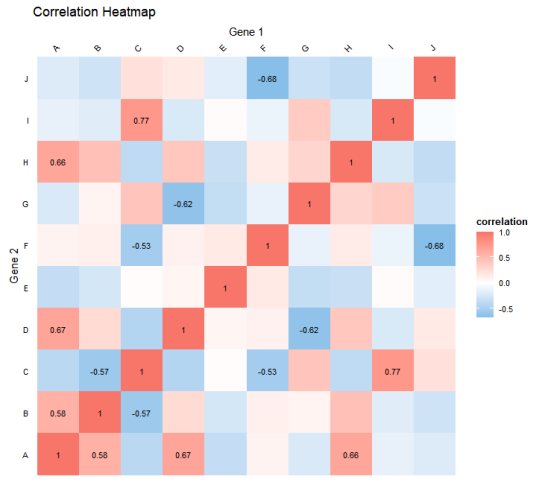
本篇文章就先到这这结束啦,后续还有其他干货文章大家点个关注不迷路哦!
本文摘自《R语言医学数据分析实践》,获出版社和作者授权发布。


















 3594
3594

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









