



传统的expression index
主要包含FPKM、RPKM、还有TPM,作为归一的工具,让不同重复、不同组别的表达量可以进行比较。
其中TPM被认为是更优的归一方法,用于映射单个基因的表达量在整体表达量中的情况。
RSEM(目前最准确的定量工具)
**RSEM ****(RNA-Seq by Expectation Maximization):**是一个转录本定量的计算工具,input file可以是FASTQ、BAM等。用于估算RNA-seq数据中每个基因和转录本的表达水平输出结果可以是read count、TPM或FPKM等多种格式,
这个计算需要基于转录本的长度但是并不是根据转录本的全长

from:26.stat115 chapter 4.6 rsem vs salmon_哔哩哔哩_bilibili
这张图可以看出在一个transcript中,两段的nt覆盖率较低,RSEM在定量过程中,会选取中间高质量的部分,从而减少bias。这也就比手动写脚本来计算表达量的方式要更优。(因为FPKM、TPM这些计算公式本身很简单,很多人可能会手动计算)
对于不同的Isoform(可变剪切产生的不同transcript)
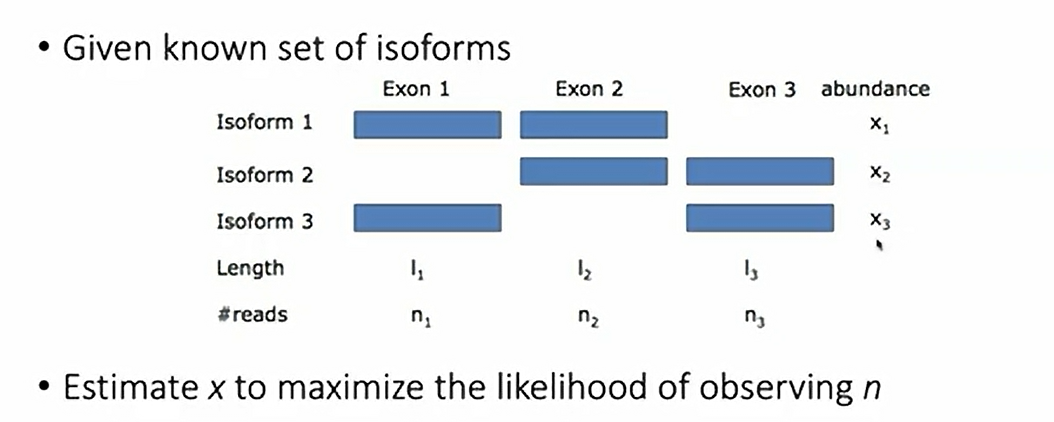
from:26.stat115 chapter 4.6 rsem vs salmon_哔哩哔哩_bilibili
RSEM还可以使用最大似然法(ML)提供精确的转录本表达量估计,考虑到每个配对到每个exon的reads来估算不同的isoform的表达量。
但是基于目前我们对转录本的了解有限,或者说有些未知的转录本没有被发现,这个表达量可能偶尔会存在一些偏误。
Salmon(运行速度更快)
虽然RSEM被认为是最精准的定量方法,但是它运行速度很慢。因为首先要map到基因组再定量,但是真正的转录区域其实并不大,像人类,只占了基因组的2%。可以通过将reads比对到转录本来进行定量,从而提高读取映射的速度。
Salmon特点:
-
采用准确且快速的定量方法
-
使用轻量级映射策略
-
计算效率更高,运行速度更快
这两种工具都能够有效地计算基因和转录本的表达量,为研究人员提供可靠的转录组分析结果。选择使用哪种工具通常取决于具体的研究需求、数据规模和计算资源。


















 3241
3241

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









