



为什么要进行序列匹配
序列匹配是理解生物序列信息、揭示生物学过程和开发治疗方法的基础。当我们测序完,获得的只是一堆ACTG的字符串的组合,我们看着这一串字符,并不会直接得到新的简介。通过序列匹配,科学家能够更好地解读生物数据,促进生物学和医学的研究,比如:
-
同源性搜索:确定一个新序列是否与数据库中的已知序列有同源关系,即它们是否来自共同的祖先。
-
相似性搜索:找到与查询序列相似的序列,即使它们没有共同的祖先,这有助于发现保守区域或功能域。
-
基因家族分析:通过序列匹配,可以识别属于同一基因家族的多个基因,并研究它们的共同特征。
-
生物标记发现:在疾病研究中,通过序列匹配可以发现特定的生物标志物,用于诊断、预后或治疗反应的预测。

使用mega11进行全局比对(Global aliment)
局部匹配(Local Alignment)
局部匹配则关注于序列中特定区域的相似性,而不是整个序列。它允许序列的其他部分不匹配,只寻找最优的局部区域对齐。局部匹配特别适用于那些只有部分区域相似的序列,或者当序列之间有大量插入、删除或突变时。
Smith Waterman算法是最常用的局部比对的算法。该算法属于动态规划,适用于部分相似序列的局部对齐。例:
胰岛素是一个调节血糖水平的激素,它在人类和许多其他哺乳动物中起着至关重要的作用。人类和鼠类是哺乳动物,它们的胰岛素基因是同源的,即它们起源于共同的祖先。
局部比对揭示了尽管两个基因的整体序列存在一些差异,但在编码胰岛素蛋白的关键区域,序列是高度保守的。保守区域通常与基因的重要功能相关。在这个例子中,胰岛素蛋白的活性位点在人类和鼠类中是高度保守的,这表明这些位点对于胰岛素调节血糖的功能至关重要。
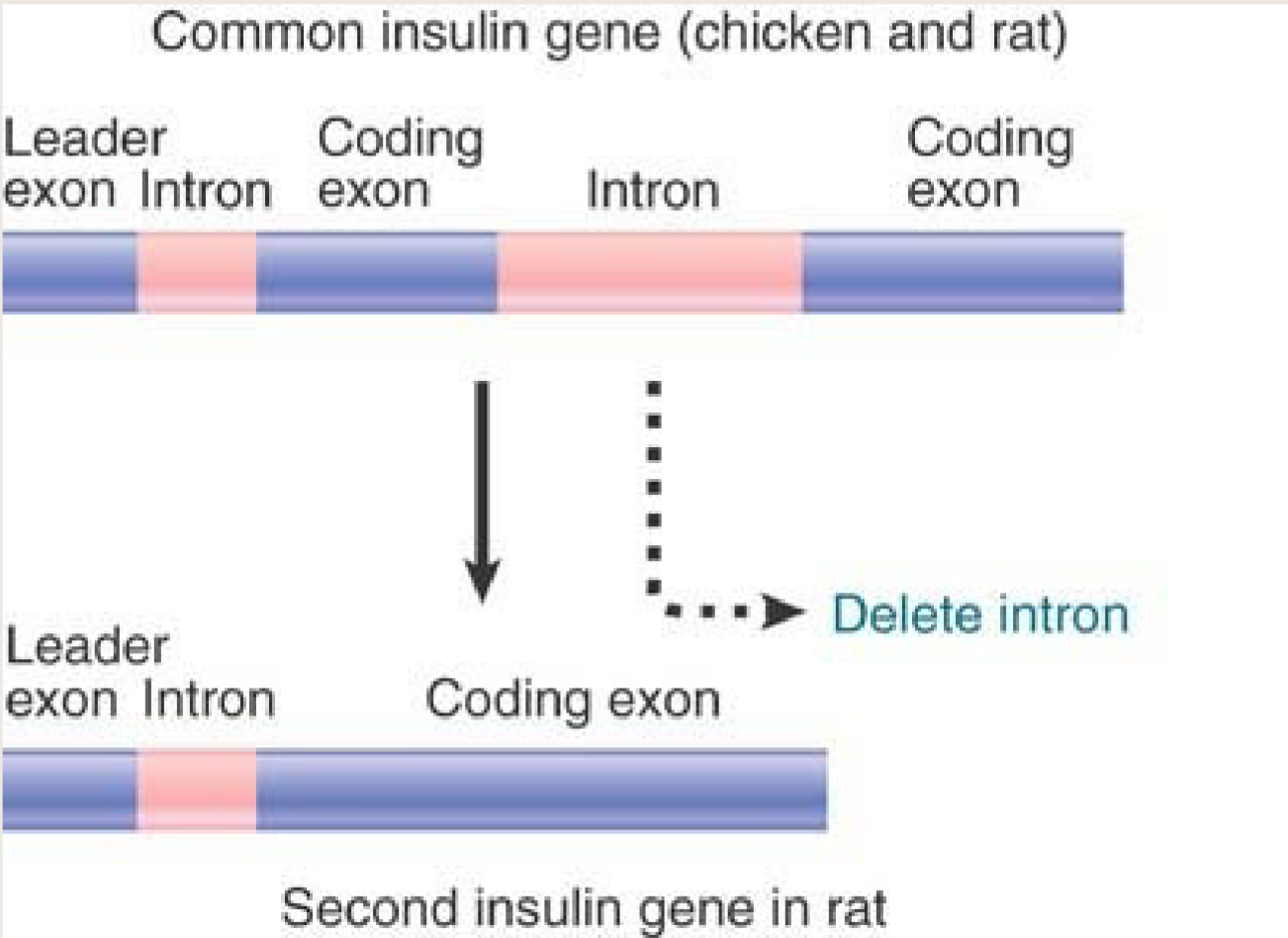
胰岛素基因在鸡、兔子和鼠中的差别。出处:《gene XII》
Smith Waterman 算法步骤
在动态规划算法中,解题思路往往是把一个问题分解成更小更简单的子问题,通过解决子问题,来得到原问题的解。
-
**初始化:**创建一个矩阵,其大小为序列A和序列B的长度加1。这个矩阵用于存储比对分数。
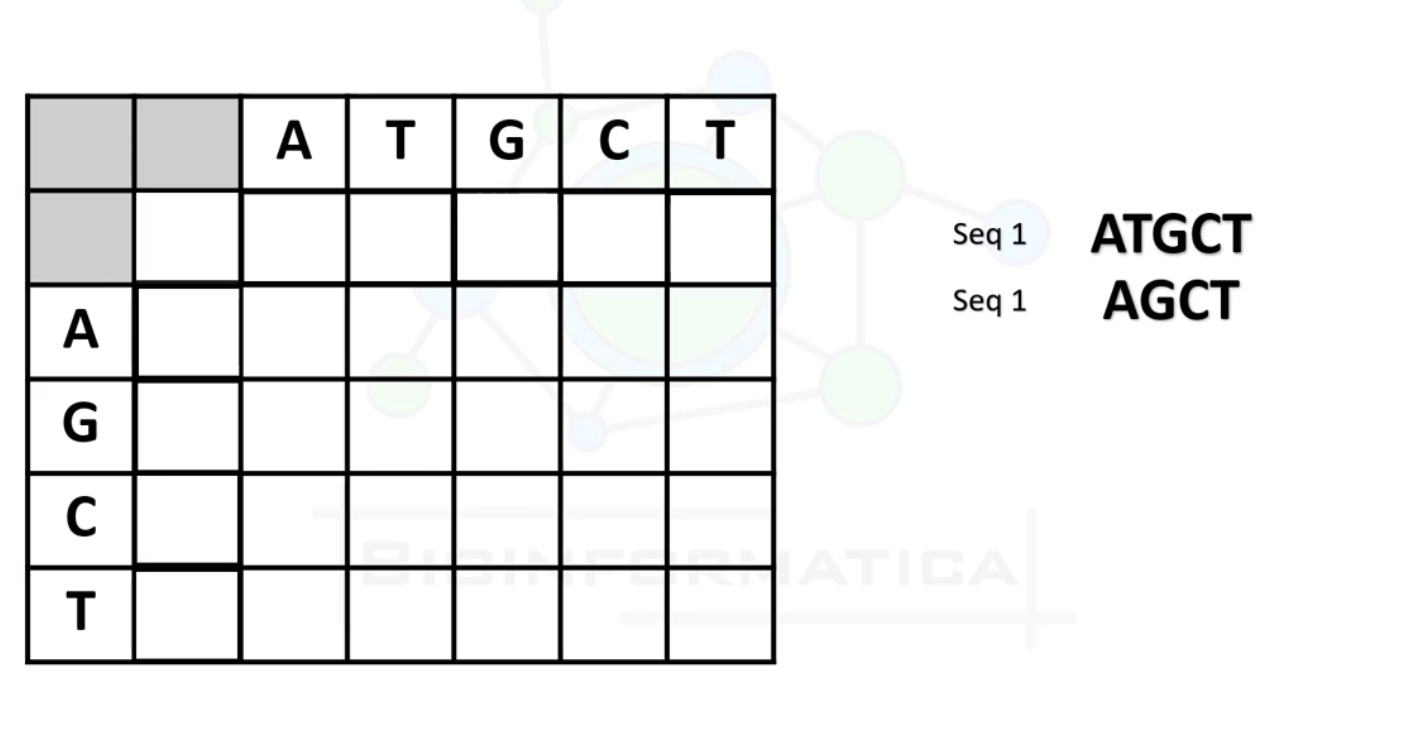
出处:视频课截图 -
得分规则:定义
匹配match得分(通常为+1),错配mismatch得分(通常为-1)和空位罚分gap(通常为-2)。 -
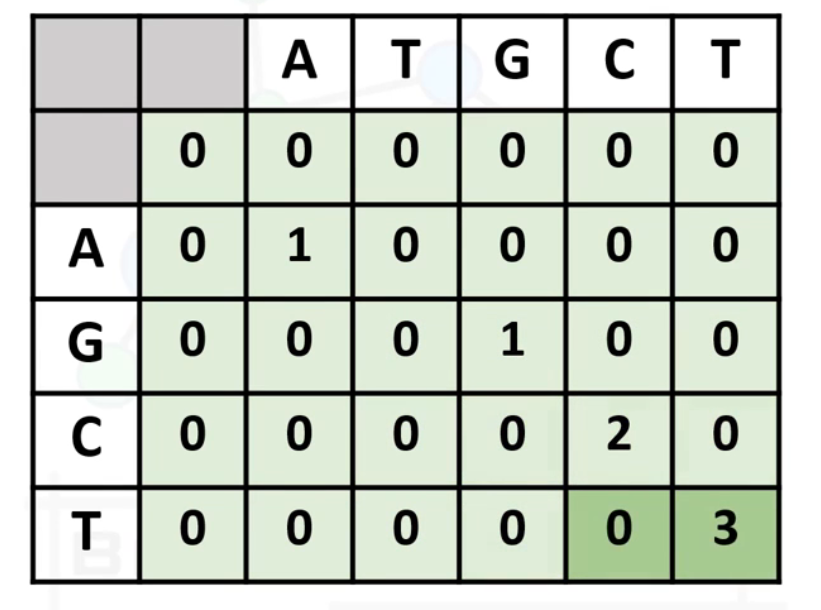
填充矩阵:从矩阵的左上角开始,逐个填充矩阵的每个格子。填充的规则如下:
- 如果当前比较的两个字符相同,则得分通常是正数(例如+1)。
- 如果字符不同,则得分通常是负数或零(例如-1或0)。
- 每个格子的得分是以下三个值中的最大值:
- 左边格子的得分-2(代表插入一个空格的惩罚)
- 上边格子的得分-2(代表插入一个空格的惩罚)。
- 左上角格子的得分+1(如果字符相同)或-1(如果字符不同),这代表两个字符匹配或不匹配的得分。
- 如果计算出的得分是负数,则将该格子设置为0,因为局部比对只关心非负的相似度得分。
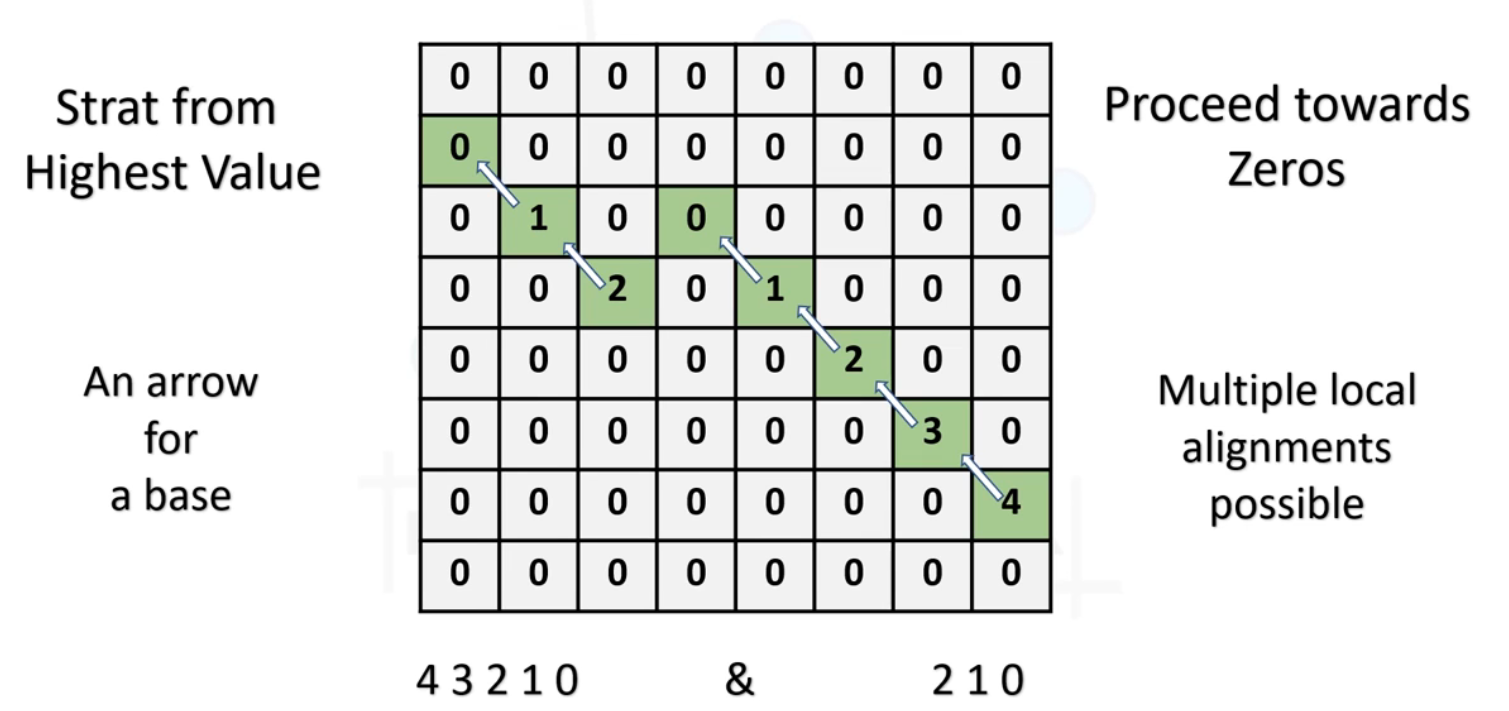
- **追踪回溯:**一旦矩阵被填充,算法通过回溯找到得分最高的路径。这条路径代表了最优的局部比对。回溯从矩阵中的最高得分点开始,沿着得分下降的路径移动,直到达到得分为0的点。


















 1437
1437

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









