



如图,从GEO上下载的数据做分析,在热图进行聚类时结果是聚类分不开,想问这个怎么解决呢
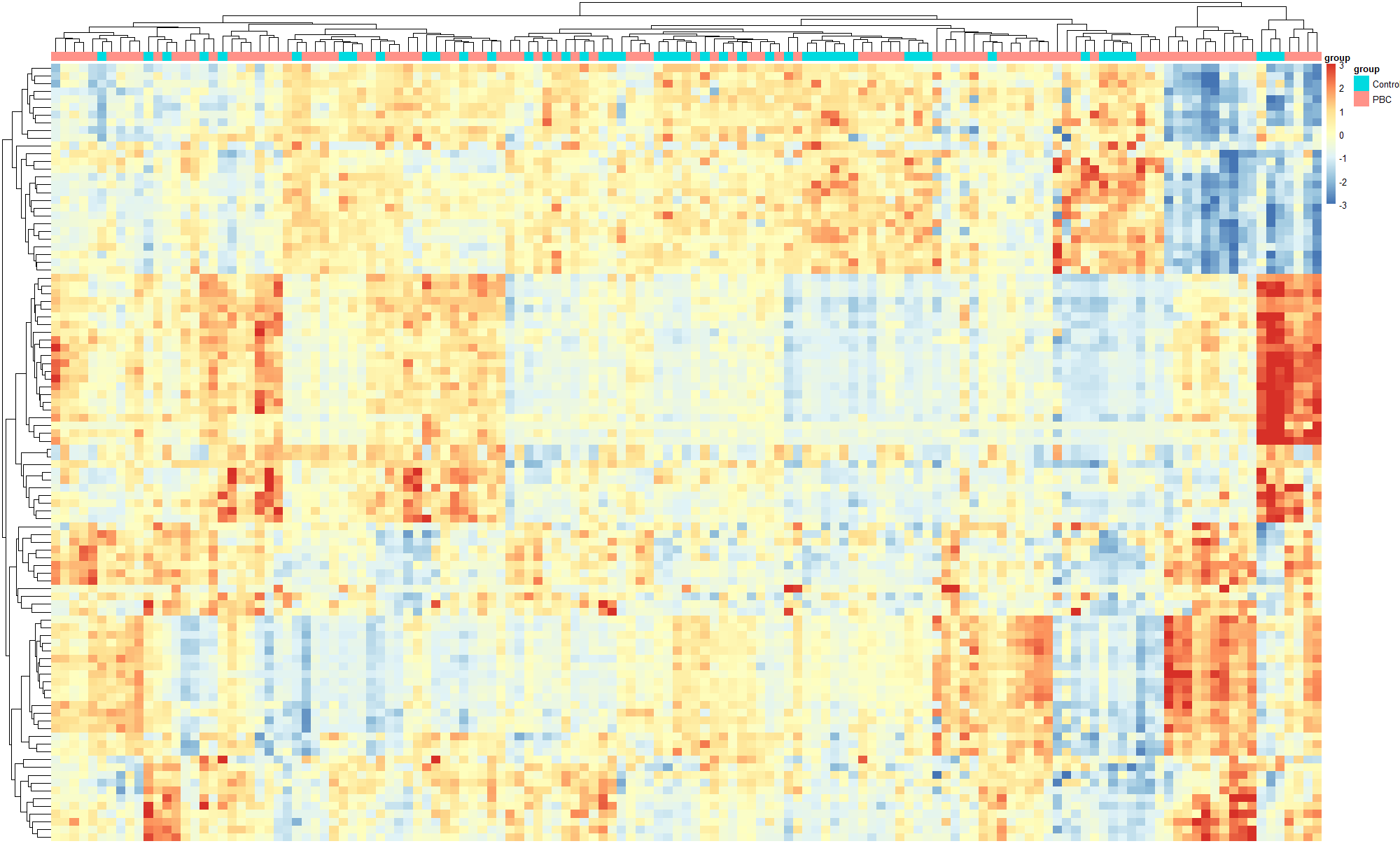
是不是因为下载的数据没有log的原因呢,从GEO上下载下来的芯片数据有些基因表达量都能有10000多,这个要先取log才能进行后续分析吗
 在对GEO下载的芯片数据进行分析时遇到聚类问题,可能由于原始数据表达量极高(如超过10000),导致聚类效果不佳。考虑数据预处理时进行log转换,这通常是解决此类问题的一种方法,以使数据分布更均匀,便于后续分析。
在对GEO下载的芯片数据进行分析时遇到聚类问题,可能由于原始数据表达量极高(如超过10000),导致聚类效果不佳。考虑数据预处理时进行log转换,这通常是解决此类问题的一种方法,以使数据分布更均匀,便于后续分析。
如图,从GEO上下载的数据做分析,在热图进行聚类时结果是聚类分不开,想问这个怎么解决呢
是不是因为下载的数据没有log的原因呢,从GEO上下载下来的芯片数据有些基因表达量都能有10000多,这个要先取log才能进行后续分析吗
 5332
5332






















 到【灌水乐园】发言
到【灌水乐园】发言






