



用于肠道常驻物种Bacteroides thetaiotaomicron的遗传回路设计自动化
信件
https://doi.org/10.1038/s41587‐020‐0468‐5
1生物工程系,麻省理工学院,美国马萨诸塞州剑桥市。2现地址:DeepBiome治疗公司,美国马萨诸塞州剑桥市。✉电子邮件:
cavoigt@gmail.com
脆弱拟杆菌是一种与人体相关的细菌,在肠道微生物组中 具有递送治疗药物的潜力1。治疗性细菌若能根据肠道内外环 境条件启动不同的基因表达程序,将大有裨益;然而,在 脆弱拟杆菌2–5中,可用于调控的元件及其组合方法一直较为 有限。我们报告了在该物种中实现Cello电路设计自动化软 件6的应用。首先,我们表征了一组基于单导RNA( CRISPR–dCas9)的基因组整合型非门/或非门,以构建用 于Cello的Bt用户约束文件(UCF)。随后,设计了可整 合响应胆汁酸和脱水四环素(aTc)传感器的逻辑电路,其 中包括一个能够区分生物制造环境、人体肠道环境以及释放 后环境的电路。该电路在实验室条件下至少12天保持稳定, 并且在体外人类肠道模型系统中与原代结肠上皮单层细胞相 关的细菌中发挥功能。
工程菌可作为“智能”治疗手段,用于递送药物或疫苗、 检测或预防感染,并响应生理变化7。尽管可以持续表达治疗 性有效载荷,但若能设计细菌以响应环境信号则更有意义; 例如,在特定微环境中触发药物释放、将治疗维持在特定水 平,或在体内开关疗法8–11。Bacteroides是革兰氏阴性专性厌 氧菌,占粪便物质中细菌总数的三分之一12–19。由于其能在哺 乳动物结肠中稳定定植并调节宿主免疫系统,因此是极具吸 引力的治疗候选载体。它们已被研究用于结肠炎和自闭症的 治疗,并已有工程菌株被设计用于改善定植和生物containment20–22。已有报道用于遗传操作的工具2–4 , 23,但如 何可预测地组合这些元件以构建更复杂的功能(包括基因回 路)仍是挑战。
定植在体内的细菌可被改造以响应原位条件、食物或水 中的小分子,并能够区分炎症、发烧、肠道出血、肿瘤和病 原体等疾病状态2 ,3,20, 24–34。已为拟杆菌属开发出小分子传感器, 可在其定植于宿主时诱导表达2,3,20。作为内源信号,胆汁酸提 供了一种识别肠道内特定区域的方法。胆汁酸的不同形式在 进入十二指肠后形成浓度梯度,并在通过肠道的过程中被细 菌化学修饰35–40。人类大肠中的胆汁酸浓度为~500 mM,其中 三分之一为脱氧胆酸(DCA)41。胆汁酸的改变在结肠癌或肝癌、遗传性疾病、肥胖、糖尿 病和炎症的发生相关36,42–44。一些致病菌利用TetR同源物来响应 胆汁酸45–47。基于细菌调节器,已在哺乳动物细胞中构建了合成 胆汁酸传感器48。
仅靠传感器只能产生简单的响应。遗传电路可以实现更复杂 的信号处理;例如,用于识别环境的逻辑、周期性药物递送、诊 断记忆,或通过通信控制毒素释放4,26,32,49,50。Cello( www.cellocad.org)的开发旨在实现电路设计自动化,并简化将合 成调控整合到基因工程项目的流程6。Cello的输入包括传感器、 期望的电路功能(以Verilog表示)以及用户约束文件(UCF), 后者指定了生物体、遗传位置、门技术及映射约束51。首个 UCF(Eco1C1G1T1)专用于Escherichiacoli NEB10β中的p15a 质粒上的电路,包含12个基于阻遏蛋白的NOR/非门。Cello利 用门的响应函数(输出启动子随输入启动子变化的方式)将它们 连接起来,以构建指定的电路。将门彼此连接或与传感器连接时, 要求其输入和输出共享一个共同信号载体,该载体被定义为 RNA聚合酶(RNAP)通量,并以相对于参考启动子的相对启动 子单位(RPUs)来衡量52。设计的电路在大肠杆菌中按预期发 挥作用;然而,无法直接将Eco1C1G1T1用于其他细菌物种。
在新宿主中实现电路设计自动化的第一步是定义一个参考启 动子,用于以RPUs表征传感器和门启动子。E. coli启动子在拟 杆菌属中效果不佳,因此我们为此目的选择了一个不同的组成 型启动子(PPAM3),并构建了参考菌株(多形拟杆菌菌株 MT768)(图1a和补充图1)2。由于绿色荧光蛋白(GFP)需 要氧气进行发色团成熟,并且仅在高表达时才能在拟杆菌属中 观察到4, ,我们选择使用荧光素酶(Nanoluc)作为我们的报告 基因53。PPAM3 的发光强度通过细胞密度进行测量并归一化(方 法)。该值被定义为RPUL = 1 ,并用于归一化其他启动子的活性 (图1b)。
接下来,我们构建了一个胆汁酸传感器。首先,我们整理了一份响 应胆汁酸的已知和推定的阻遏蛋白列表,包括CmeR(空肠弯曲菌)和 BreR(Vibrio cholerae),以及六个推定的调控蛋白(VFA0359、 BCAS0007、Bamb6005、VFA0088、Smlt2112和Xcc0233)45,47,48,54,55。 基于每个阻遏蛋白,通过将基因置于组成型启动子的控制下构建了传感 器(图1c)。输出启动子的设计是在组成型启动子的−33和−7区域之间 插入操纵序列用于肠道常驻物种Bacteroides thetaiotaomicron的 遗传回路设计自动化 武谷真央1,2,张建波1,张书怡1,亚历山大·J·特里亚西1,黄宇嘉1,琳达·G·格里菲斯1和 克里斯托弗·A·沃伊特1 ✉ 自然生物技术 | www.nature.com/naturebiotechnolo gy 信件Nature BiotechNology (PcfxA)。传感器被整合到attBT1‐1中。细胞在厌氧条件下于TYG (胰蛋白胨–酵母提取物–葡萄糖)培养基中生长,在有无62.5 μMDCA的情况下培养6小时,然后测量启动子活性(补充图2和 方法)。基于VFA0359的传感器表现出优异响应,具有低基础活 性、440倍动态范围以及在8μMDCA时达到半最大值(图1d和 补充图3)。该传感器对其他胆汁酸的响应也进行了评估,发现 DCA> LCA> CDCA> CA(补充图4和补充表1)。对于结合型胆 汁酸(TCA和GCA)未观察到响应(未显示)。
我们测试了VFA0359胆汁酸传感器与先前为多形拟杆菌开发 的aTc和IPTG(异丙基‐β‐d‐硫代半乳糖苷)传感器之间的串扰。 每个传感器被分开设计,使阻遏蛋白以及输出启动子和报告基因 整合到多形拟杆菌基因组的不同位置。所有三种阻遏蛋白均由P BT1311组成型启动子转录,表达盒插入attBT‐1位点。输出 启动子(PBA、PLacO23和P1TDP)驱动Nanoluc的表达,并整合到基因 组中的一个(DCA)或两个(IPTG和aTc)位点。这些传感器对 DCA、IPTG和aTc的响应进行了评估,未观察到串扰现象(图1e和 补充图5–7)。阻遏蛋白与启动子之间也无串扰(补充图5–7)。
基因回路可以通过组合执行更简单逻辑操作的门控来构建。我们选 择将门技术针对双输入或非门功能,该功能具有两个输入启动子和一个输 出启动子。此前,我们使用蛋白质阻遏蛋白实现反相功能56,但 我们发现当将其转移到多形拟杆菌基因组时,其动态范围不足 (未显示)。作为替代方案,我们采用CRISPR干扰技术,利 用小向导RNA(sgRNA)引导失活的Cas9(dCas9)抑制靶 启动子(图1f)。此方法此前已被我们及他人用于在大肠杆菌 和酿酒酵母2 ,57–60中构建遗传逻辑。尽管在大肠杆菌 58,中表达 dCas9时观察到毒性,但在多形拟杆菌中未观察到生长影响 (补充图8)。
每个门控基于不同的单导RNA。单导RNA通过随机化DNA 识别序列,并通过计算扫描基因组以确保其不存在而设计(方法)。 可抑制启动子通过将相应的目标序列放置在PBF2884的不同位置并 筛选最高抑制效果而设计,最高抑制发生在−7位置上游(PMsite3) (补充图9)。当添加62.5 μMDCA以诱导单导RNA转录时,导 致发光强度降低20倍(方法)。另外设计了多个单导RNA以创建 包含七个门控的正交集合(补充图10、11)(方法)。尽管可以 构建更大的文库,但我们发现在大肠杆菌中,超过7个以上的单导 RNA无法在单个电路中使用,否则会显著消耗dCas9资源,并使 门响应降低至<10‐fold58,61。
门控需要强终止子以避免通读至其他门控或染色体基因。 此外,每个门控应使用不同的终止子,以避免重复的DNA序 列在构建或携带电路时引起重组。在拟杆菌属中,终止子尚未 得到充分表征,因此我们从大肠杆菌文库中选择了多个强终止 子,并设计了一个系统来量化其在多形拟杆菌中的强度(方法 和补充图12)。
非门基于单导RNA、启动子和终止子构建。dCas9的表达 由组成型P1启动子控制(图1g和补充图13)。通过增加DCA 浓度并测量门控输出启动子在RPU中的响应,来测定每个门控 的响应函数L (图1h和补充图14、15)(方法)。然而,为了 预测如何连接门控,输入也必须以RPUL表示,而非DCA浓度。 为校正响应函数,构建了一个独立菌株(多形拟杆菌菌株 MT772)以测量PBA启动子活性随DCA的变化情况,这些数 据用于校正响应函数的x轴(补充图14)。校正后的响应函数 被拟合为数学形式,并从中提取参数(补充表2)。
通过添加第二个输入启动子,可将非门转换为或非门。为 避免串联启动子可能引起的阻滞现象23,我们选择通过复制一份 sgRNA(及其相关的终止子),并将每份分别置于不同的输入 启动子控制下来构建或非门(图1i和补充图16,17)。或非门 具有二维的响应函数(图1j),但如果能基于其一维的非门响 应函数(图1h)并使用输入启动子活性之和来简化设计自动化 算法,则更为简便。实验设计用于检验这种方法是否足够准确, 结果表明非门响应函数确实可以被使用(补充图16)。
DNA序列、响应函数的参数化以及每个门控的生长影响(以 600nm处的光密度(OD600)测量)被整合,用于构建针对多形 拟杆菌的UCF(Bth1C1G1T1,补充文件1)。指定生物体为多形 拟杆菌VPI-5482,其attBT2‐1和attBT2‐2位点含有一个表达盒, 用于dCas9的组成型表达。UCF中包含Eugene规则,规定门控 元件应位于输出基因上游的正向位置(补充图18)。最终或门通过拆分最后一个启动子以驱动两个拷贝的输出基因(例 如,nanoluc)来实现。UCF规定电路应插入到attBT1‐1中。
Cello被用于设计能够整合胆汁酸和aTc传感器的逻辑电 路。逻辑操作可以在Verilog中指定,通过所需的真值表或具 体的连线图来实现。运行后,算法会优化sgRNA在每个门上 的分配,并组装序列以形成编码该电路的线性DNA。系统可 预测在所有输入组合(aTc和DCA的存在与缺失)条件下输出 基因的表达情况。由于响应函数基于荧光素酶测量结果,因此 它们代表的是细胞群体平均值。
首先,我们设计了一个实现异或逻辑的电路:当且仅当其 中一个输入开启时,输出开启。根据真值表,用Verilog编写 (图2a),逻辑最小化算法识别出最优的连线图(图2b)。然 后,利用sgRNA门控的响应函数计算连线图中每个位置的最 佳sgRNA。该搜索通过模拟退火算法进行,旨在最大化应关 闭的最高输出与应开启的最低输出之间的差异(电路评分) (图2c)。接着,使用Eugene规则将该分配转换为线性 DNA序列(图2d)。预测响应显示为图2e中的彩色虚线。需 要注意的是,这些预测是在电路构建之前做出的。含有该电路 的多形拟杆菌菌株严格按照设计构建。测量了其对不同诱导剂 组合的响应,发现与预测值高度吻合(图2e)。
治疗性细菌若能在不同阶段开启不同的基因表达程序,将 大有裨益。这可以通过多输入多输出逻辑实现,该逻辑整合一 组传感器以识别环境。作为原理验证,我们设想设计一种电路, 使其响应对应于发酵罐条件(有诱导剂但无胆汁酸)、肠道环 境(有胆汁酸但无诱导剂)以及治疗后排出至体外环境(无诱 导剂且无胆汁酸)的信号(图3a)。每种条件被分配一个不同 的输出启动子,从而可在各条件下控制目标基因的表达。所需 的逻辑用Verilog编写(图3b),Cello据此设计出相应的电路 和DNA序列(图3c,d)。为了使用相同的报告基因测量输出, 构建了三种菌株,将每个输出启动子连接到nanoluc。需要注 意的是,这三个菌株具有相同的电路和全部三个输出启动子, 但对于未被测量的启动子,其对应的nanoluc位置被替换为无 效DNA序列(图3e)。电路的输出在代表4种输入组合和3种 输出的12种状态中均与预测结果高度吻合(图3f)。
为了评估我们电路的长期遗传稳定性,菌株在厌氧室中培 养了12天,并以随机的方式在状态间切换。在此生长实验期间, 细胞并未被严格维持在指数期,有时会在厌氧室中放置较长时 间。这样做的目的是为了对细胞施加压力,并使其经历不同的 生长条件和营养水平。在此期间,电路表现符合预期,未出现 任何损坏迹象(图3g)。电路从关闭到开启以及从开启到关闭 的特征时间约为6小时。
人胃肠道模型63被用于在多形拟杆菌细胞黏附于人上皮单 层的背景下测试电路功能。覆盖肠表面的单层通过原代结肠干 细胞类器官忠实地再现,这些类器官可分化为吸收性、杯状细 胞和肠内分泌细胞(图4a)(方法)63。与源自癌细胞系的模 型不同,黏膜层由杯状细胞分泌,更准确地反映了多形拟杆菌 自然黏附的人上皮涂层(图4b)64。黏附后,多形拟杆菌在其 转录组中发生全局性变化,寻找替代碳源,并生长减缓64。上 皮层细胞分泌的分子(如抗菌肽)也会影响多形拟杆菌的转录 组65。使用该胃肠道模型使我们能够在这一细胞和环境背景的 真实模型中对电路性能进行原型验证。
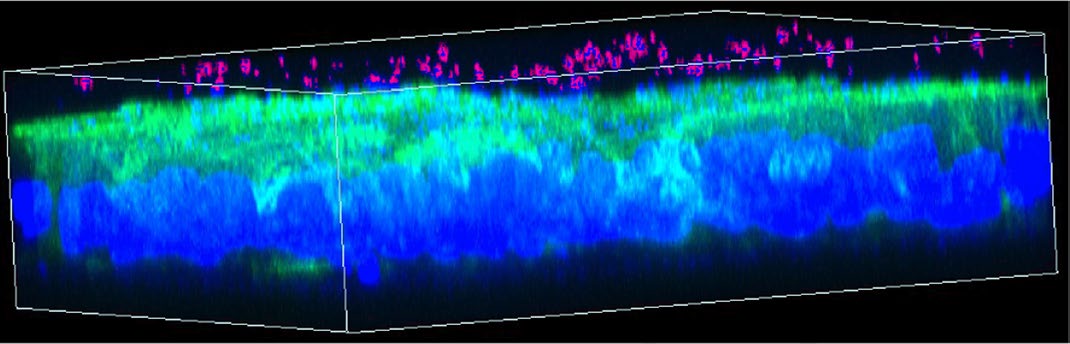
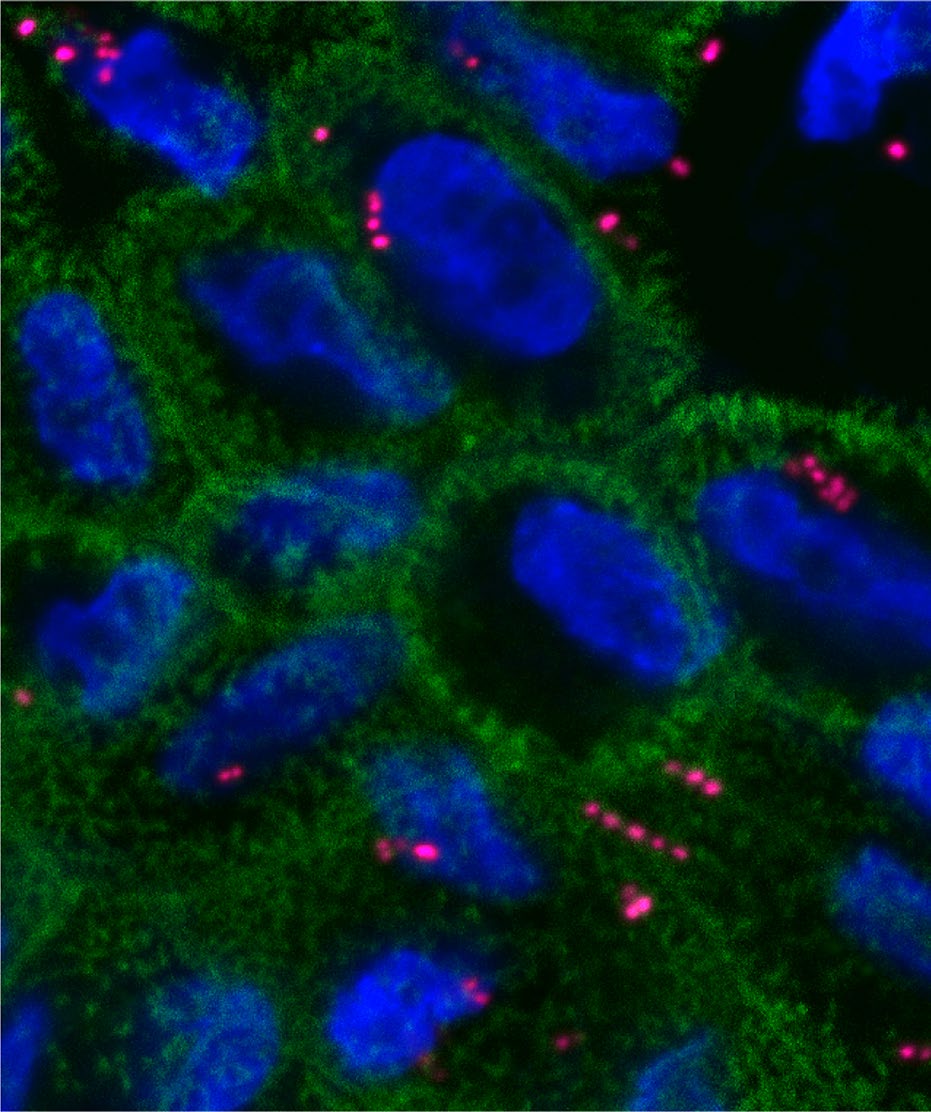
使用胃肠道模型评估双输入三输出逻辑电路的电路性能。 上皮单层培养在Transwell小室上,基底侧含有支持哺乳动物 细胞生长的培养基(图4a)。评估电路时,需要测试其对多 种诱导剂组合的响应。为此,Transwell系统是理想选择,因 为可以在12孔板中建立相同的上皮单层,并在相同条件下测试 电路响应。将干细胞接种于涂有胶原的Transwell膜上,并分 化约1周(图.4a,b)。另取多形拟杆菌的过夜培养物,重悬于 新鲜预还原酵母胨脂肪酸(YCFA)培养基中,取500μl加入 到单层的顶侧并培养2小时(方法)。DAPI染色和肌动蛋白丝 免疫染色表明,多形拟杆菌细胞定位于顶侧(图.4c,d和补充 图21,22)。通过明场图像显示无可见孔洞,且跨上皮电阻 (TEER)值处于‘紧密’胃肠道上皮范围内,证实单层结构 完整(补充图23)66。
细菌黏附后,通过添加含有适当DCA和aTc组合的新鲜培 养基来诱导基因回路。细胞在含有诱导剂的条件下培养6小时, 然后通过移液去除非黏附细胞。多形拟杆菌从顶端室底部收集与单层密切相关的细胞,方法是移除培养液顶 部的450微升,然后将剩余的50微升与450微升新鲜培养液混合 培养基。其光密度和发光强度的测量方式与TYG培养样品相同。 如图4e所示,该电路的输出与体外实验结果高度一致(图3f)涵盖了代表4种输入组合和3种输出的12种状态(图4e)。
我们创建了一个用于设计多形拟杆菌基因回路的灵活平台, 并通过设计一个整合了响应胃肠道健康信号和小分子药物传感 器的电路,证明了该平台的有效性。尽管UCF仅有7个门控, 但这些门控可用于构建约1012个电路,并可连接至其他用户指 定的传感器。电路输出为报告基因,但该输出也可替换为编码 治疗性蛋白的基因,或构建化学结构复杂的抗生素或免疫调节 剂的基因簇。迄今为止,遗传回路设计自动化仅在模式生物的 实验室菌株(E. coli)以及基于质粒的系统上实现6。在此,我 们证明,将电路设计自动化扩展到遗传工具相对较少的新物种 时,并不需要对宿主进行广泛的表征。多个电路使用Cello构 建,尽管它们被设计为在特定培养条件下发挥作用,但发现其 功能正常在代表人体肠道复杂性和独特性的真实条件下进行原型设计时, 电路能够正确运行。我们的人体胃肠道模型能够快速评估电路 的多种状态,但代价是仅能研究8小时内的电路响应,之后细 菌过度生长会破坏单层结构。更精细的微生理系统可用于评估 电路长达数周67,68。此外,可通过增加上皮层的复杂性来研究 特定现象,例如隐窝中定植细菌的响应、蠕动和剪切应力的作 用、分子向血液的转运,以及与肠‐脑轴的相互作用。按照特 定要求设计电路并随后在真实条件下快速构建原型的能力,将 有助于推动活体响应性人类疗法的发展。

用于肠道常驻物种Bacteroides thetaiotaomicron的遗传回路设计自动化
菌株、质粒与培养基
使用的菌株为多形拟杆菌VPI-5482野生型(ATCC 29148)。骨架载体pNBU1‐erm和pNBU2‐tetQ分别由T. Lu(麻省理工学院)和M. Fischbach(斯坦福大学)提供。所有多形拟杆菌菌株均在TYG肉汤或脑心浸液(BHI;BD BBL BHI琼脂编号211065)琼脂中培养,并添加10%马血(Hemostat DHB500)。每升TYG肉汤包含:10 g胰蛋白胨(BD Bacto编号211705)、5 g酵母提取物(BD Difco编号210929)、2 g葡萄糖(Fisher编号50‐99‐7)、100 ml 1 M KPO4 (pH 7.2;将1升1 M二碱式磷酸盐(VWR编号0705‐500G)溶于H2O中,与~430 ml 1 M单碱式磷酸盐(Sigma 795488‐500G)溶于H2O中混合,调节至pH 7.2)、40 ml TYG盐溶液(含0.5 g MgSO4·7H2O(USB公司编号18651)、10 g NaHCO3 (Fisher Scientific S233)、2 g氯化钠(Amresco X190),溶解于1升H2O中)、1 ml 0.8% CaCl2 (Sigma编号C1016‐500G)水溶液、1 ml 0.4 mg ml−1 FeSO4 (Amresco编号0387‐500G)水溶液、4 ml 0.25 mg ml−1刃天青(Sigma编号R7017‐1G)水溶液、1 ml组氨酸‐血红素溶液(1.2 μg ml−1血红素:将12 mg血红素(Sigma编号H3281‐1G)溶解于10 ml 0.2 M组氨酸(Sigma编号H7875‐25G)水溶液中,调pH至8,过滤除菌)、0.5 g L‐半胱氨酸(Sigma编号30089‐25G),以及1 ml 1 mg ml−1甲萘醌(Sigma编号M5625‐25G)溶于100%乙醇中。L‐半胱氨酸使用前以水溶解(每25 mg L‐半胱氨酸加1 ml水),并立即通过0.22 μM滤膜(Pall PN4192)过滤除菌。组氨酸‐血红素溶液、甲萘醌和L‐半胱氨酸在接种前加入已灭菌的培养基中。除非另有说明,培养基在接种前需进行预还原处理(置于厌氧室中过夜)。根据需要添加抗生素:红霉素(25 μg ml−1;Acros Organics编号114‐07‐8)、四环素(2 μg ml−1;Goldbio编号64‐75‐5)和庆大霉素(200 μg ml−1;Goldbio编号1405‐41‐0)。针对多形拟杆菌的抗生素仅用于接合后的筛选,不用于常规生长。以下诱导剂浓度根据需要使用,除非另有说明:500 μM异丙基‐β‐D‐硫代半乳糖苷(IPTG)(Goldbio编号367‐93‐1)、100 ng ml−1 aTc(Acros Organics编号13803‐65‐1)和62.5 μM DCA(Sigma编号D2510‐10G)。其他使用的胆汁酸包括:胆酸(CA)(Sigma编号C9282‐25G)、鹅脱氧胆酸(CDCA)(Sigma编号C8261‐1G)、石胆酸(LCA)(Sigma编号L6250‐10G)、牛磺胆酸(TCA)(Sigma编号T4009‐1G)和甘氨胆酸(GCA)(Sigma编号G7132‐1G)。大肠杆菌S17-1 λ pir(来自T. Lu)或EC100D pir(VWR编号75927‐934)用于扩增基于NBU1和NBU2的构建体,在37 °C有氧振荡条件下培养于LB(BD Difco编号244620)肉汤或LB琼脂(BD Bacto编号214010)中,并添加100 μg ml−1羧苄青霉素(Goldbio编号4800‐94‐6)用于转化后的筛选和生长。
接合转移至多形拟杆菌及基因组编辑
基于pNBU1和pNBU2质粒的构建体(补充图24)通过接合转移整合到拟杆菌属基因组中,接合所用菌株为E. coli S17-1 λ pir。pNBU1基于一种可移动的拟杆菌转座子NBU1,含有intN1,该基因介导pNBU1上的attN1位点与多形拟杆菌基因组中attBT1‐1位点(位于tRNA‐Leu基因3′端)的特异性重组。pNBU1包含一个突变的attN1,其中−2位点由G变为C,已证明该突变可提高整合频率。将E. coli S17-1 λ pir与多形拟杆菌的过夜培养物按1:1比例混合于TYG培养基中,并点样于含+ 10% 马血的BHI血琼脂平板上。接合反应在需氧条件下于37°C孵育。>16小时后,刮取菌体,并连续划线接种至添加了庆大霉素和红霉素(针对pNBU1)或庆大霉素和四环素(针对pNBU2)的BHI血琼脂平板,于37°C厌氧条件下孵育。24–48小时后出现的菌落通过使用基因组和载体特异性引物进行PCR验证其是否发生位点特异性整合。当需要将pNBU2和pNBU1质粒同时整合入多形拟杆菌时,先进行pNBU2的接合转移,再进行pNBU1。本研究所构建的所有菌株均列于补充表3中,其质粒图谱见补充图24。
厌氧生长
所有多形拟杆菌菌株均在厌氧室(Coy实验室;乙烯基厌氧室A型手套箱型号7000000)内的强制通风培养箱(Coy实验室Model 2000型号8543025)中于37 °C静置培养。厌氧室中的氧气通过钯催化剂(Coy实验室型号6501050)持续去除,该催化剂每周更换一次,方法是在90°C烘箱中孵育过夜或更长时间。氮气(Airgas型号NIUHP300)用于清除气闸室内的空气,并维持厌氧室内的正压。厌氧室内的氧气和氢气水平使用CAM‐12厌氧监测仪(Coy实验室型号6250000)进行监测。除非打开气闸室的内门以将物品移入或移出,否则厌氧室内的氧气维持在0百万分率;在此情况下,会有低水平的氧气进入室内,但会迅速下降。为确保钯催化剂正常工作,通过添加5% H2, 20% CO2, N2 气体混合物(Airgas定制混合气,型号X03NI75C2O05502)使室内氢气浓度保持在1.5%以上。在将物品移入或移出时,使用默认的P1程序(2次真空和吹扫气体(N2)循环,随后1次真空和混合气体循环)。
荧光素酶检测和RPUL
将多形拟杆菌在TYG培养基中过夜培养的菌液用新鲜TYG培养基1:100稀释,并取180微升多形拟杆菌培养物的等分试样在光密度(OD)600介于0.4至0.7之间(约6小时)时取样。从该培养物中取15 μl与15 μl Nanoluc反应缓冲液(Promega Nano‐Glo 荧光素酶系统 N1120;底物按1:50比例加入)混合。使用Synergy H1 多功能微孔板读板机(BioTek 型号 8041000)测量光密度(OD)600和发光强度。对于发光检测,增益设置为100,检测时间为1秒。两项测量均在96孔平底微量滴定板(Nunc 型号 165305)上进行。相对发光单位(RLU)每菌落形成单位(CFU)通过将原始发光值除以CFU计算得到,CFU根据绘制OD600与菌落形成单位(CFU)的标准曲线并通过OD600 值计算得出。为了将荧光素酶活性转换为RPUL,定义了一个RPUL标准菌株(多形拟杆菌菌株MT768)(图1a,b)。通过将待测样品的发光强度(RLU每CFU)除以RPUL标准菌株的发光强度(RLU每CFU),将其转换为RPUL。详见生命科学报告摘要。
传感器表征
编码CmeR、BreR和BreR同源蛋白的DNA使用Jcat针对多形拟杆菌VPI-5482进行密码子优化,并合成为gBlocks(IDT)。含有传感器的多形拟杆菌培养物被连续划线接种到BHI血琼脂平板上。24小时后挑取单菌落,接种至14毫升Falcon离心管(Fisher编号14‐959‐11B)中的2毫升TYG(未预还原)培养基中,并在厌氧室中于37°C静置孵育。次日将过夜培养物以1:100稀释,在PlateOne深孔96孔2毫升聚丙烯板(USA Scientific 1896‐2000)中加入800微升预还原TYG培养基,分别添加或不添加相应的诱导剂,静置培养6小时。6小时后取180微升等分试样,测量荧光素酶活性和OD600(如上所述)。传感器串扰实验采用相同方法进行。化学串扰的诱导倍数通过计算诱导培养物的发光强度(RLU/CFU)与未诱导培养物的比值得出。当用作图坐标轴标签时,“抑制(−阻遏蛋白/+阻遏蛋白)”指缺乏阻遏蛋白的菌株的归一化发光强度(RLU/CFU)除以含有阻遏蛋白的菌株测得的归一化发光强度(RLU/CFU)所得的比值。
终止子表征在多形拟杆菌中
测量含有表达盒的菌株以及在启动子和开放阅读框之间插入终止子的菌株的发光强度(RLU/CFU)(补充图12)。将含有终止子的多形拟杆菌甘油菌种在BHI血琼脂平板上连续划线接种。24小时后挑取单菌落,接种至14毫升Falcon离心管(Fisher编号14‐959‐11B)中的2毫升TYG(未预还原)培养基中,并在厌氧室内静置孵育。次日将过夜培养物按1:100稀释,在PlateOne深孔96孔2毫升聚丙烯板(USA Scientific 1896‐2000)中加入800微升预还原TYG培养基,有或无相应诱导剂,静置培养6小时。6小时后取180微升等分试样,测定荧光素酶活性和OD600(如上所述)。
sgRNA设计
使用随机DNA序列生成器(http://www.faculty.ucr.edu/~mmaduro/random.htm;GC含量概率参数为0.5)生成包含TANNTTTG的20 bp随机DNA序列,并通过BLASTn对该序列与多形拟杆菌VPI‐5482基因组进行比对,以查找基因组中可能受影响的脱靶位点。未发现任何20核苷酸序列在基因组内与前间隔区序列邻近基序(PAM)相邻位点具有同源性。
sgRNA正交性
构建了7个sgRNA和7个sgRNA‐操纵子启动子的所有可能组合,并接合转移至多形拟杆菌菌株MT724中,共获得49个菌株(多形拟杆菌 MT809–MT857)。含有sgRNA与sgRNA‐操纵子启动子配对的多形拟杆菌甘油菌种在BHI血琼脂平板上连续划线接种。24小时后挑取单菌落,接种于14 ml Falcon离心管(Fisher编号14‐959‐11B)中的2 ml TYG(未预还原)培养基中,在厌氧室内静置孵育。次日将过夜培养物以1:100稀释,于96深孔板(USA Scientific 1896‐2000)中加入800微升预还原TYG培养基,静置培养6小时,同时添加或不添加相应的诱导剂。培养6小时后取180微升等分试样,测定荧光素酶发光强度和OD600(方法如前所述)。通过将未诱导培养物的发光强度除以诱导后多形拟杆菌培养物的发光强度,计算抑制倍数。
门表征
门测量质粒、RPUL 标准以及胆汁酸传感器(用于将DCA浓度转换为输入启动子活性)均接合转移至多形拟杆菌菌株MT724中。含有门控、RPUL 标准和胆汁酸传感器的多形拟杆菌甘油菌种在BHI血琼脂平板上连续划线接种。24小时后挑取单菌落,接种于14毫升Falcon离心管(Fisher编号14‐959‐11B)中的2毫升TYG(未预还原)培养基中,并在厌氧室内静置孵育。次日将过夜培养物以1:100稀释,于PlateOne深孔96孔2毫升聚丙烯板(USA Scientific 1896‐2000)中,在一系列DCA浓度(62.5, 37.5, 22.5, 13.5, 8.1, 4.9, 2.9, 1.7, 1.0, 0.6, 0.4, 0.1)条件下,使用800 μl预还原TYG培养基静置培养6小时。收集等分试样(180 μl)在6小时后采集样本,并测量荧光素酶和OD600。使用RPU绘制输入启动子相对于输出启动子的活性,并通过Prism的非线性回归工具对数据进行希尔函数拟合,所得拟合参数见补充表2。输入阈值“IL”(输入低)和“IH”(输入高),用于确定两个非门之间的连接性,初始计算分别为最大输出的0.5倍和最小输出的2倍,如先前所述6。然而,对于Bth1C1G1T1 UCF,将IL调整为0.001,IH调整为0.2,以增加可找到的电路设计解决方案的数量。
使用Cello的电路设计
Cello软件和代码可免费获取(https://github.com/CIDARLAB/cello)。Cello通过Cello API在本地运行,生长评分阈值设置为0.75。使用了一个UCF文件(Bth1C1G1T1.UCF.json)(补充文件1)。应用Eugene规则将门控映射到线性DNA序列。为了将输出门分配至3′位置,Eugene中包含规则,使得所有逻辑门均在输出门前分配(在Eugene中写作‘gate_M1 BEFORE gate_nanoluc’)。逻辑门的顺序为随机,这是Eugene中的默认选项。每个转录单元的方向设置为相同且全部正向(在Eugene中写作‘ALL_FORWARD’)。无aTc时使用0.03 RPUL,100 ng ml−1 aTc时使用6.4 RPUL,无DCA时使用0.01 RPUL,62.5 μM DCA时使用1.5 RPUL。
表征电路在时间进程中的行为
挑取含有RPUL和双输入三输出电路的多形拟杆菌菌株(多形拟杆菌 MT798、MT799和MT800分别对应报告输出2、3和1)的单菌落,接种于不含诱导剂的TYG(未还原)培养基中过夜培养。第二天,在厌氧室内(VWR Galaxy微型离心机)将1 ml培养物离心,沉淀用含aTc的预还原TYG清洗两次,并以1:50比例稀释至800微升含相同培养基的TYG中。接种至新的输入状态后,立即按描述每2小时一次,持续8小时进行OD600和发光强度测量。8小时后,将培养物置于厌氧室中37°C过夜。第二天,在厌氧条件下将培养物离心,并再次用对应下一输入状态(仅DCA)的含诱导剂预还原TYG清洗两次,然后以1:50比例稀释至含相同培养基的TYG中。同样在输入状态转换后立即每2小时一次,持续8小时进行OD600和发光强度测量。此过程对所有可能的排列重复进行。
原代结肠上皮单层培养基
原代结肠上皮单层细胞使用基础培养基(DMEM/F12(Gibco,12634‐010 500 ml)添加5 ml 谷氨酰胺–I(Gibco,35050‐061)、5 ml HEPES(Gibco,15630‐080)和5 ml 青霉素‐链霉素(Gibco,15140‐148))、类器官生长培养基(Wnt、R‐脊椎蛋白、诺金蛋白(由波士顿儿童医院(D. 布雷尔特)提供的WRN条件培养基)、基础培养基以及1X B‐27补充剂50× (Gibco,17504‐001)、1X N‐2补充剂100× (Gibco,17502‐001)、10 mM 烟酰胺(Sigma‐Aldrich,N0636)、500 μM N‐乙酰半胱氨酸(Sigma‐Aldrich,A9165)、10 μM Y‐27632二盐酸盐(Biogems,1293823)、10 μM SB202190(Biogems,1523072;Tocris,1264)、500 nM A 83‐01(Biogems,9094360)、50 ng ml−1小鼠EGF(PeproTech,AF‐315‐09)、人[Leu15]‐胃泌素I(Sigma‐Aldrich,G9145)和5 nM 前列腺素E2(Biogems,3632464))、接种培养基(接种培养基成分:65% WRN条件培养基,32.5% 基础培养基,1X B‐27补充剂50×,1X N‐2补充剂100×,10 μM N‐乙酰半胱氨酸,5 μM SB202190, 2.5 μM 噻唑维林,500 nM A 83‐01, 50 ng ml−1小鼠EGF,人[Leu15]‐胃泌素I,5 nM 前列腺素E2)和分化培养基(分化培养基成分:20% R‐脊椎蛋白1条件培养基,80% 基础培养基(无抗生素),1X B‐27补充剂50×,1X N‐2补充剂100×,500 μM N‐乙酰半胱氨酸,500 nM A 83‐01, 50 ng ml−1人诺金蛋白(PeproTech,120‐10C),50 ng ml−1小鼠EGF,以及人[Leu15]‐胃泌素I。
肠道上皮单层
结肠单层模型源自原代人结肠干细胞类器官63。类器官的建立与维持按照科赫研究所和麻省理工学院Yilmaz实验室先前描述的方法进行69。在马萨诸塞州总医院,经供体知情同意后,采集一名去标识个体(30 岁憩室炎患者)直肠乙状结肠部位内镜组织活检的外观正常区域。实验方法遵循科赫研究所机构审查委员会和麻省理工学院人类实验受试者使用委员会的指导方针。类器官在Matrigel滴中培养,并每7天以1:3比例传代。传代后第4天进行换液。为制备单层,于第7天将类器官收集至15 ml锥形管中。通过离心(1,000g × 5 min,4 °C)形成类器官沉淀,随后通过抽吸去除培养基。然后加入细胞回收溶液(Corning,354253;每100 μl Matrigel加1 ml)以破坏类器官沉淀,接着将类器官悬浮液冰上孵育45–60分钟。之后将类器官悬浮液再次离心沉淀,并用1 ml预热的PBS−/− (Gibco,10010‐023)重悬,其中含有2.5 mg/ml胰蛋白酶(Sigma,T4549)和0.45 mM EDTA(Ambion,AM9260G)。重悬的类器官在37°C水浴中放置5分钟,然后使用带弯头吸嘴的1000微升移液器手动解离为单细胞。细胞用台盼蓝和自动细胞计数仪(英潍捷基)进行计数。将细胞用接种培养基稀释至浓度为每毫升60万个细胞,取500 微升加入到每个12孔包被Transwell的顶侧(表面积:1.12 cm2),并在基底外侧加入1.5毫升无细胞接种培养基。在接种前,Transwell在培养箱中用鼠尾胶原I(吉布科,A10483‐01,50 μg ml−1溶于磷酸盐缓冲液)包被1‐2小时,然后在加细胞前立即用磷酸盐缓冲液洗涤。
原代结肠上皮单层细胞与多形拟杆菌的共培养
使用分化后的单层细胞与多形拟杆菌进行共培养。接种后第3天,通过在顶侧更换为无抗生素基础培养基,在基底外侧更换为分化培养基,使单层细胞完成分化。细胞分化后4‐5天的分化单层用于上皮细胞–多形拟杆菌共培养。多形拟杆菌细胞的培养方法如前所述,但将TYG培养基或BHI琼脂平板替换为YCFA培养基(AS‐680,厌氧系统公司)或YCFA琼脂平板(AS‐675,厌氧系统公司)。未使用TYG培养基,因为初步实验表明单独使用TYG培养基会破坏单层结构。多形拟杆菌细胞重悬于500微升不含诱导剂的新鲜YCFA中,然后加入到单层细胞的顶侧。在加入多形拟杆菌细胞之前,吸除顶侧培养基。随后立即把单层细胞转移至培养箱(5%二氧化碳,37°C)。孵育2小时后,向培养基中加入诱导剂,并继续孵育6小时。6小时后,移除450微升顶部顶侧培养基,取50微升含有沉降到上皮细胞表面的细胞的培养基,稀释后与450微升新鲜YCFA混合。按上述方法测定OD600和发光强度。收集多形拟杆菌后,通过加入500微升磷酸盐缓冲液,并将Transwell放入连接至EVOM2上皮电压计的EndOhm室中测量经上皮电阻。参见生命科学报告摘要。
免疫荧光染色与成像
使用4%甲醛固定单层细胞10分钟。为了阻断非特异性结合,将固定后的细胞在BlockAid封闭液(赛默飞B10710)中于轻柔振荡(50转/分钟)条件下孵育1小时。随后,细胞在封闭液中加入偶联鬼笔环肽(ab176753)和DAPI进行染色,并在铝箔覆盖下于4°C过夜孵育。次日用磷酸盐缓冲液清洗两次,每次5分钟。然后将带单层细胞的膜从Transwell小室中取出,置于玻片上,滴加抗荧光淬灭剂后加盖薄圆形盖玻片。使用蔡司LSM 880共聚焦显微镜进行成像。三维图像通过软件Zen 2.3 SP1 FP3,版本14.0.20.201(蔡司)进行重建。明场图像使用EVOS细胞成像系统拍摄。对于图4c,d,使用GIMP‐2.10将图像中的细菌从DAPI蓝色伪彩色显示为粉红色,以突出显示相对于哺乳动物细胞的细菌。亮度和对比度也已增强。
报告摘要
有关研究设计的更多信息,请参见与本文关联的《自然科研报告摘要》。
数据可用性
遗传元件和UCF文件Bth1C1G1T1可作为补充信息获取。以下质粒的DNA序列已存入GenBank:pMT405(MN991273);pMT406(MN991274);pMT423(MN991275);pMT444(MN991276);pMT445(MN991277);pMT447(MN991278);pMT448(MN991279);pMT449(MN991280);pMT450(MN991281);pMT451(MN991282);pMT455(MN991283);pMT462(MN991284);pMT468(MN991285);pMT469(MN991286);pMT470(MN991287);pMT492(MN991288);pMT493(MN991289);pMT494(MN991290)。支持本研究发现的数据可向通讯作者在合理要求下获取。
代码可用性
Cello软件和代码可免费获取(https://github.com/CIDARLAB/cello)。
参考文献
69 Roper, J. et al. 结直肠癌和转移的体内基因组编辑和类器官移植模型。自然生物技术 35, 569–576 (2017)。
致谢
We thank M. Mimee (MIT) 感谢T. Lu(麻省理工学院)提供了基于NBU1的Bacteroides穿梭载体pNBU1‐Erm。感谢M. Fischbach(斯坦福大学)提供了基于NBU2的Bacteroides穿梭载体pNBU2‐tetQ。原代人结肠上皮细胞由O. Yilmaz(麻省理工学院)实验室慷慨提供。感谢K. Schneider(麻省理工学院)和C. Wright(麻省理工学院)提供的技术协助。本工作得到了美国国立卫生研究院P50资助(P50‐GM098792)和海军研究办公室多学科大学研究计划项目(N00014‐13‐1‐0074)、国防高级研究计划局协同发现与设计(SD2;FA8750‐17‐C‐0229)、国家科学基金会用于信息处理和存储技术的半导体合成生物学(SemiSynBio;CCF‐1807575)项目以及美国国立卫生研究院(NIH R01EB021908)。
利益冲突
C.A.V.和M.T. 已基于本工作提交了临时专利申请。其余所有作者均无利益冲突。
附加信息
补充信息可在 https://doi.org/10.1038/s41587-020-0468-5 获取。通信和材料请求应联系 C.A.V.重印和权限信息可在 www.nature.com/reprints 查询。

















 1826
1826

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









