



有时候设计引物跑不同样本,不确定是否有产物使用
→primer-blast
填入引物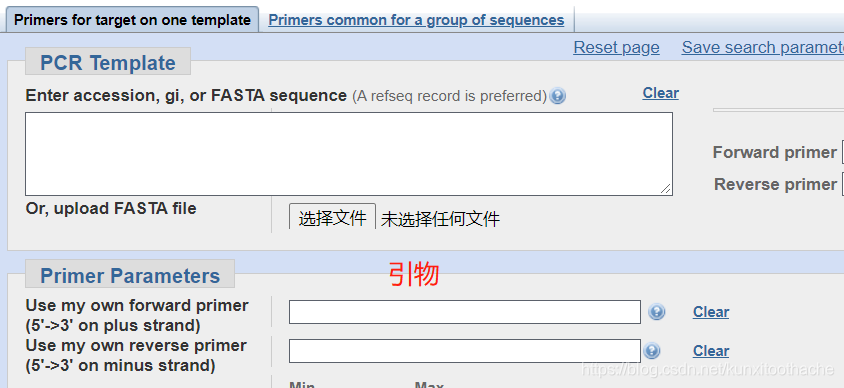
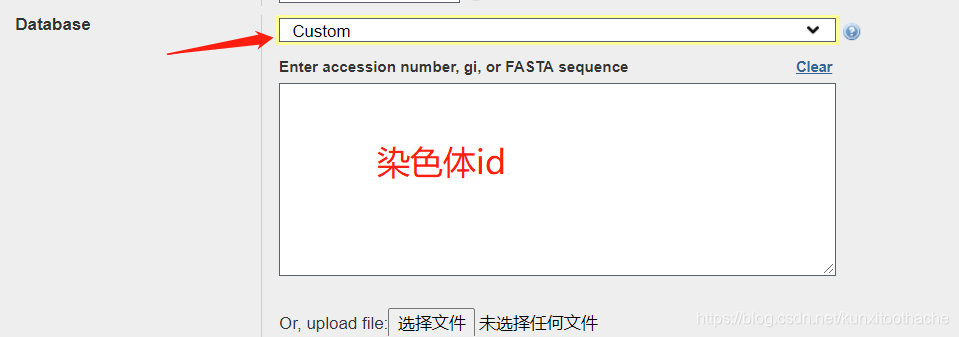
设置选custom,填入目标染色体id
do it

NCBI查找目标染色体是否有目标产物
最新推荐文章于 2021-12-09 21:50:07 发布
有时候设计引物跑不同样本,不确定是否有产物使用
→primer-blast
填入引物
设置选custom,填入目标染色体id
do it
 1万+
1万+

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言


























