



 这篇博客介绍了如何利用Multiwfn程序处理lammps输出的分子动力学轨迹文件,展示体系中的孔洞和自由区域。通过详细步骤,包括lammps的dump指令、pdb格式转换、Multiwfn的后处理设置,以及不同参数对结果的影响,最终确定最佳参数组合,以实现可视化效果与计算速度的平衡。
这篇博客介绍了如何利用Multiwfn程序处理lammps输出的分子动力学轨迹文件,展示体系中的孔洞和自由区域。通过详细步骤,包括lammps的dump指令、pdb格式转换、Multiwfn的后处理设置,以及不同参数对结果的影响,最终确定最佳参数组合,以实现可视化效果与计算速度的平衡。
背景:由于课题需要,之前所用的后处理程序是同门师姐给的框架,根据自身体系进行修改内容,但计算出的结果是数值,而不是可视化的直观样式。那天同门师兄给我推荐了个链接说可能用得到。最近一些事情耽搁了下,现在按操作流程处理并记录。
该篇内容是默认你已经具备了基础的分子动力学知识和检索学习的能力(即:如有疑问请百度~)
步骤:
1. lammps运行输出原子坐标(这里以体相EmimNTF2的离子液体为例)
dump 1 all custom 20000 dump.lammpstrj id mol type element x y z ix iy iz
dump_modify 1 sort id element CR NA CW CM CA CT HR HW HM HT NBT SBT OBT CBT F1
2. 处理输出的轨迹文件(dump.lammpstrj)为pdb格式(Multiwfn可识别的其它格式也可)
从服务器上下载dump.lammpstrj到本地,用Notepad++打开,选取一帧里完整的内容到Excel
(刚刚输出的轨迹文件格式如下,复制带坐标行的即可)


选中复制的数据,在菜单栏依次点击数据-分列-下一步-空格-完成,将所选数据按空格分开
再将有用的原子名称和坐标单独复制到新建的xyz文件中,仅保留原子的元素名(ctrl+alt按列选中后下拉删除,再按需要补齐空格)

保存成.xyz文件格式,用VESTA打开,转换成.pdb格式(File-Export Data-选中PDB格式)
3. Multiwfn后处理(有一些小细节需要注意,我也是尝试了2h左右才摸清楚一些问题的原因)
启动Multiwfn程序,即双击安装包里的这个.exe文件
![]()
拖入刚刚转换得到的.pdb文件 ,拖入后会显示路径(路径中不要有中文,否则闪退)
![]()
无误后回车
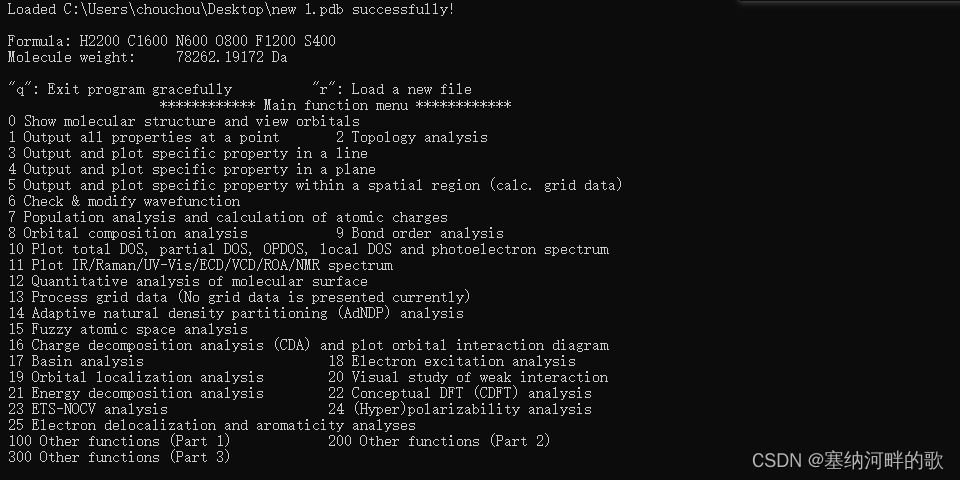
输入:300(//其它功能(Part 3)),回车
-----------------------------------------------------
输入:1 (//观看自由区域和计算自由区域体积),回车
输入:4 (//修改用于平滑边界的展宽函数),回车
输入:1 (//Gaussian展宽),回车
输入:1.8 (//用原子范德华半径的1.8倍作为Gaussian函数的半高全宽(FWHM)),回车
【这里教程上是1.8,我也不清楚为什么,但是我之前的后处理程序是直接用的范德华半径,因此这里也可输入1,不过对比了下两个参数对于结果的影响,发现自由体积分数的数值一致但可视化的图不一样,我目前还不太明白】
原子范德华半径值:原子的范德华半径(单位nm),用里面的Bondi[2]即可
-----------------------------------------------------
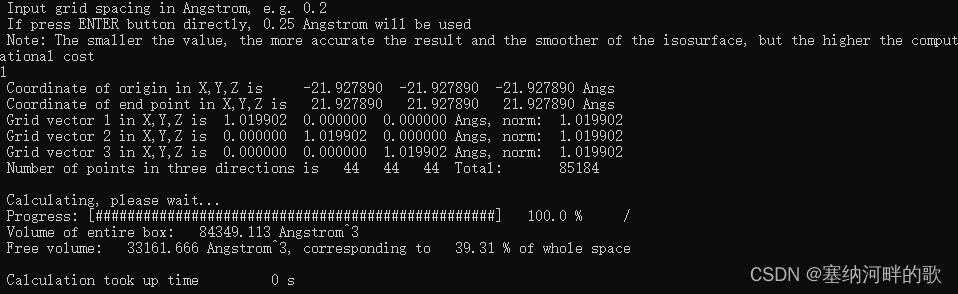
输入:1 (//设置格点并开始计算),回车
输入: -21.92789,-21.92789,-21.92789(输入轨迹文件中的值,且必须用英文标点符号,否则闪退) (//如果直接回车,则默认是原点位置,即(0,0,0)处,这和当前情况不一致)
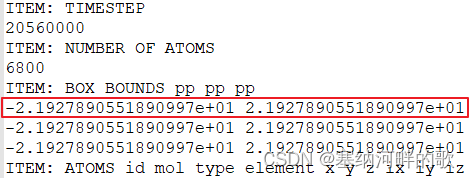
输入:43.85578,43.85578,43.85578 (输入轨迹文件中的值的两倍)(//如果直接回车,则默认用晶胞的三个边长作为格点数据计算范围的三个边长,但这里的pdb文件中没有边长,不修改的话最后自由体积分数就会是0)
输入:1 (//如果直接回车,则默认用0.25埃格点间距。格点间距越小,要算的格点数就越多,耗时就越高,图像也会越平滑,因此应根据实际情况决定。当前格点间距下的图像质量已经不错了)
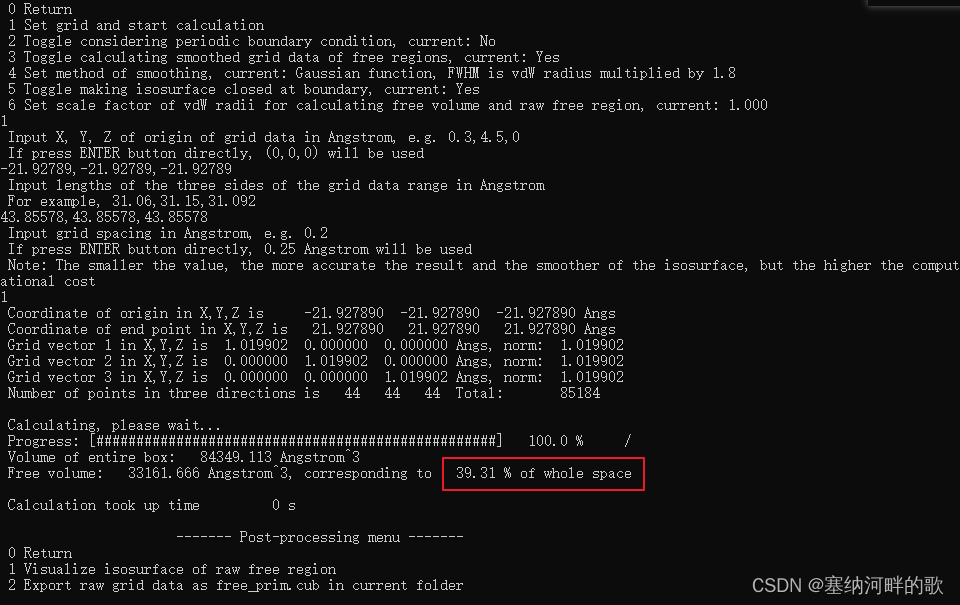
算完后可以看到
Volume of entire box: 84349.113 Angstrom^3
Free volume: 33161.666 Angstrom^3, corresponding to 39.31 % of whole space
这说明整个盒子总体积,即三个边长的乘积是 84349.113 Angstrom^3,自由体积是33161.666 Angstrom^3,占总体积的 39.31%。
自由体积是这样算的:对于每个格子,如果它距离任意一个原子的距离在此原子的范德华半径以内,这个格子就被认为是占据的。所有没有占据的格子数乘上格子体积就是自由体积。
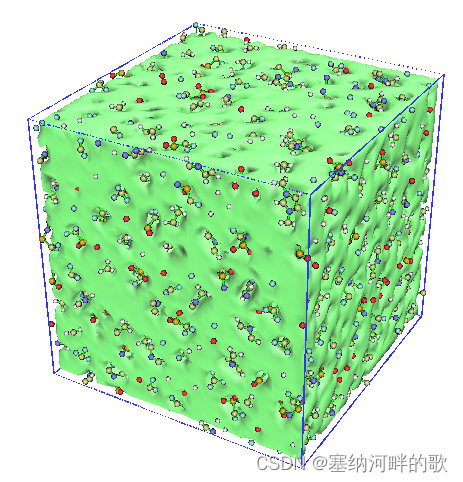
输入:3-回车,就出现了图形界面,将窗口右侧的Show molecule和Show data range都选上,可看到带框的图片。

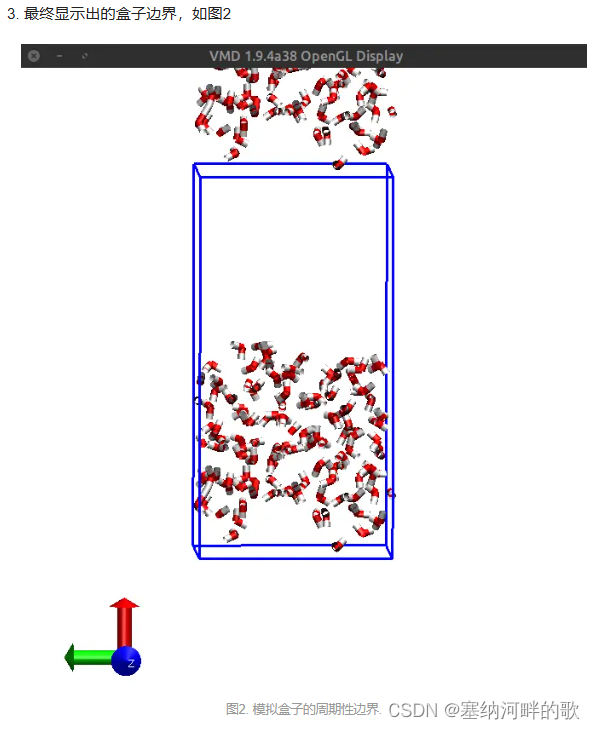
上图中的等值面直观地展现出了盒子中未被原子占据的区域
4. VMD后处理
为了得到更好的图像效果,点击Return后,选择4,将格点数据导出为当前目录(Multiwfn安装目录)下的free_smooth.cub文件

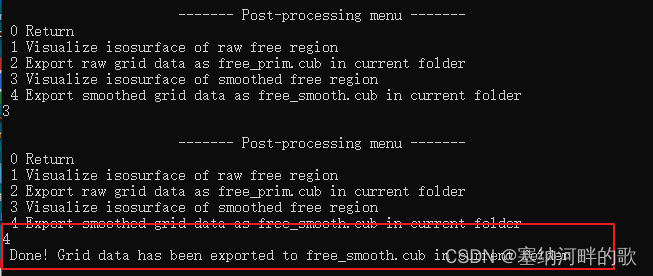
![]()
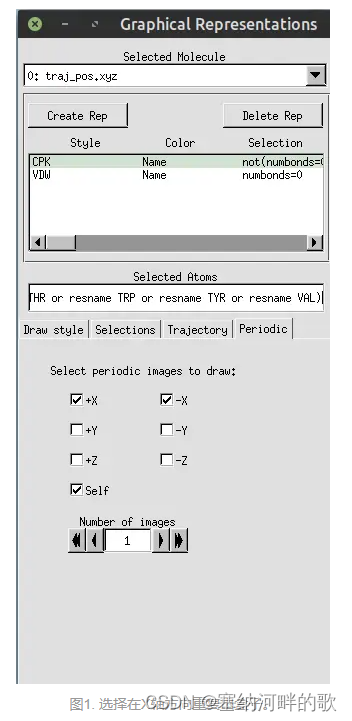
启动VMD后将之载入,进入Graphics - Representation,点Create Rep,把设置改为下面的样子来显示出等值面
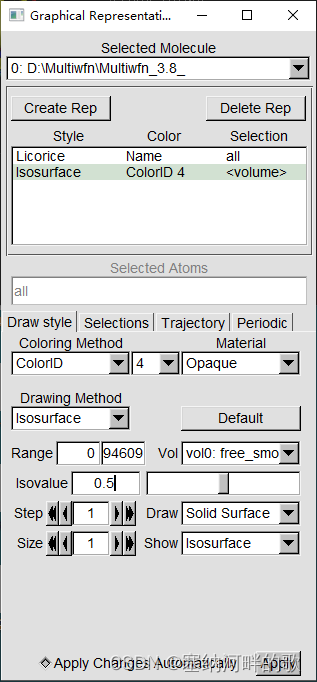
Graphics-Colors:
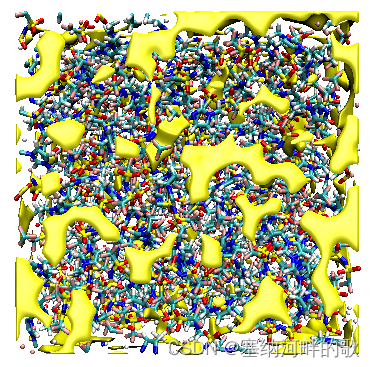
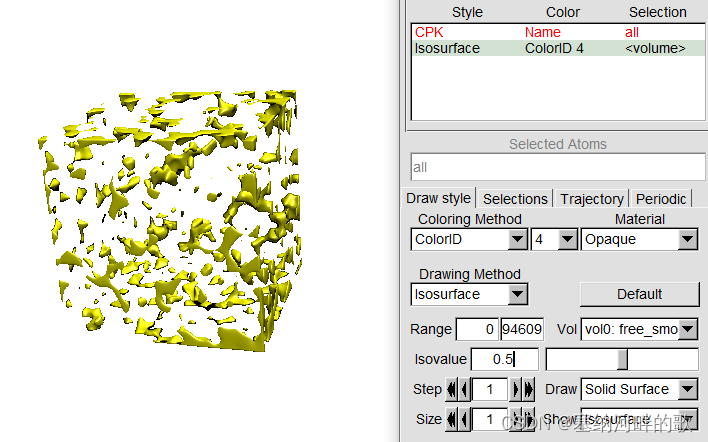
在命令窗口输入:pbc box -center origin (加一个盒子)

其余有用的命令:如何在VMD中画出模拟盒子

5. 参数对比
使用参数:原子范德华半径的1.8倍+1埃格点间距=39.31%(计算速度非常快)
Volume of entire box: 84349.113 Angstrom^3
Free volume: 33161.666 Angstrom^3, corresponding to 39.31 % of whole space
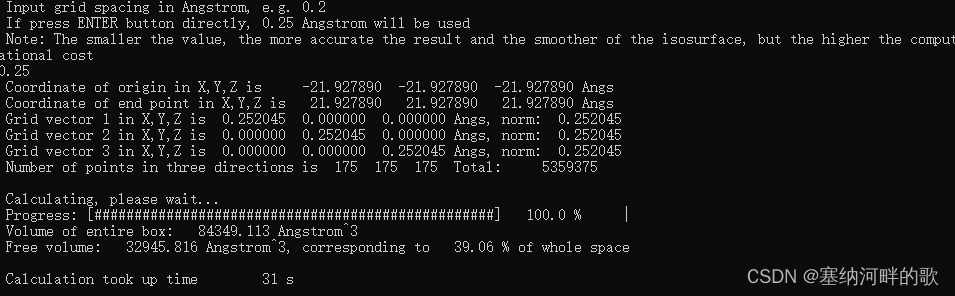
使用参数:原子范德华半径的1.8倍+0.25埃格点间距=39.06%(计算大概30s)
Volume of entire box: 84349.113 Angstrom^3
Free volume: 32945.816 Angstrom^3, corresponding to 39.06 % of whole space

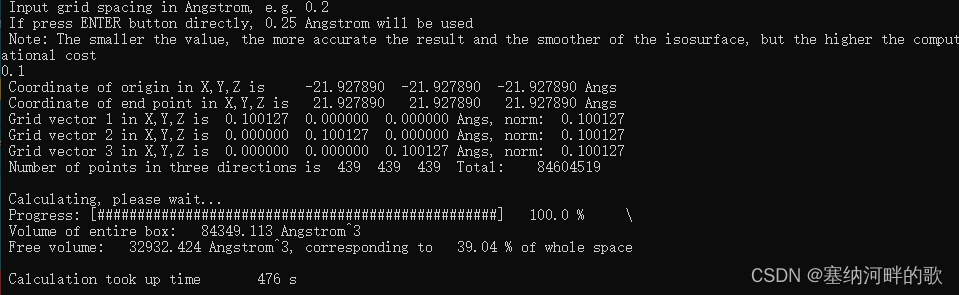
使用参数:原子范德华半径的1.8倍+0.1埃格点间距=39.04%(计算大概8min)
Volume of entire box: 84349.113 Angstrom^3
Free volume: 32932.424 Angstrom^3, corresponding to 39.04 % of whole space

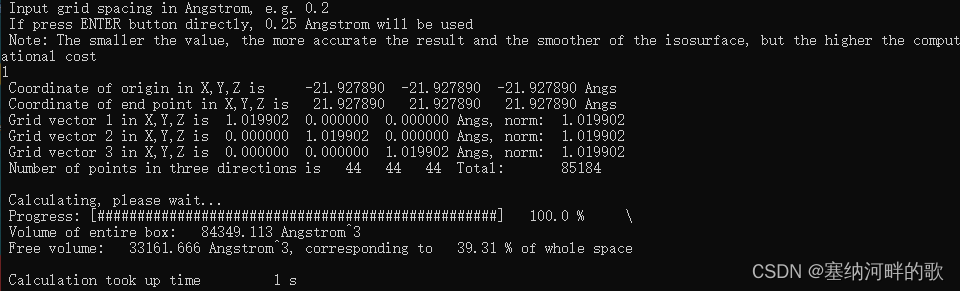
使用参数:原子范德华半径的1.0倍+1埃格点间距=39.31%(计算速度非常快)
Volume of entire box: 84349.113 Angstrom^3
Free volume: 33161.666 Angstrom^3, corresponding to 39.31 % of whole space
使用参数:原子范德华半径的1.0倍+0.5埃格点间距=39.12%(计算速度快)
Volume of entire box: 84349.113 Angstrom^3
Free volume: 32996.044 Angstrom^3, corresponding to 39.12 % of whole space
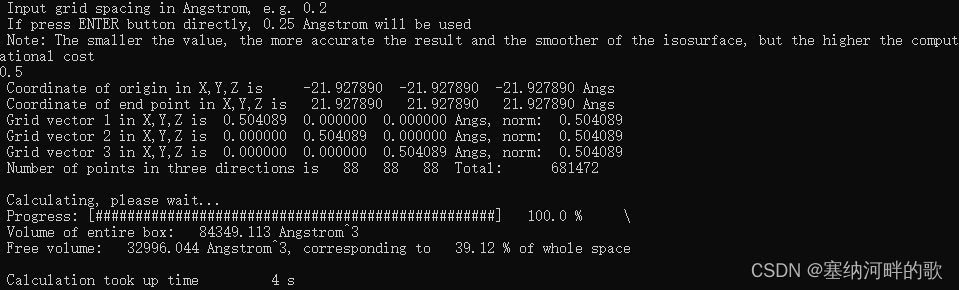

使用参数:原子范德华半径的1.8倍+0.25埃格点间距=39.06%(计算大概30s)
Volume of entire box: 84349.113 Angstrom^3
Free volume: 32945.816 Angstrom^3, corresponding to 39.06 % of whole space
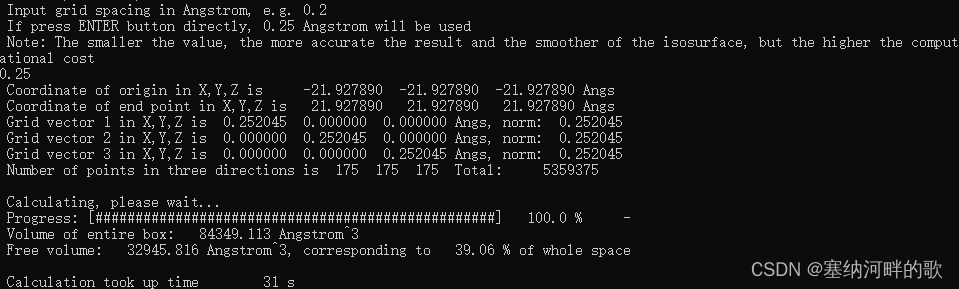

对比完后发现最优组合确实是教程上的默认值:原子范德华半径的1.8倍+0.25埃格点间距
【最优标准:可以达到计算目的即可视化明确,同时运算速度可接受】


















 1284
1284

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









