






WGCNA(Weighted Gene Co-expression Network Analysis)是一种用于分析基因共表达网络的方法。在WGCNA中,与基因表达量相关的一些关键概念包括模块(module)、模块特异性基因(eigengene)以及与模块和基因相关的一些统计量,如ME(Module Eigengene)、KME(eigengene-based connectivity)、hME(in-module connectivity)等。
-
ME(Module Eigengene):
-
定义: 模块特异性基因(eigengene)是模块内所有基因的主成分,它代表了整个模块的表达模式。ME是模块特异性基因的一个值,用于表示模块整体的表达情况。
-
理解: ME可以看作是对整个模块在不同条件下的表达进行综合评估。ME的变化可以帮助理解模块在不同生物学条件下的调节情况。
-
-
KME(eigengene-based connectivity):
-
定义: KME是指每个基因与模块特异性基因(ME)之间的相关性。在WGCNA中,通过计算每个基因与模块特异性基因的相关性,可以评估基因在模块内的连接性。
-
理解: KME提供了关于每个基因在模块内位置的信息,即基因与整个模块的联系紧密程度。高KME值的基因在模块内可能具有更重要的调控作用。
-
-
hME(in-module connectivity):
-
定义: hME是指每个基因在模块内与其他基因的连接性的平均值。它表示了一个基因在其所在模块中的相对中心性。
-
理解: hME反映了一个基因在模块内的相对重要性。具有高hME的基因可能在模块内具有较强的连接性,可能在模块内发挥关键作用。
-
这些统计量通常与基因的表达模式和调控性质相关。通过WGCNA,研究人员可以识别基因模块,并进一步研究这些模块与生物学特性之间的关系。ME、KME和hME等指标提供了对基因在模块内位置的不同度量,帮助解释基因调控网络中的重要性和相互关系
链接:https://www.jianshu.com/p/fa038701c698
一、模块特异性基因 (ME)
hdWGCNA 包括一个计算单个细胞中模块特征基因的函数。
# need to run ScaleData first or else harmony throws an error:
#seurat_obj <- ScaleData(seurat_obj, features=VariableFeatures(seurat_obj))
# compute all MEs in the full single-cell dataset
seurat_obj <- ModuleEigengenes(
seurat_obj,
group.by.vars="Sample"
)二、特征基因 (hME)
ME 矩阵存储为矩阵,其中每行是一个单元格,每列是一个模块。可以使用 该函数从 Seurat 对象中提取此矩阵,该函数默认检索 hME。
# harmonized module eigengenes:
hMEs <- GetMEs(seurat_obj)
# module eigengenes:
MEs <- GetMEs(seurat_obj, harmonized=FALSE)三、模块连接度 kME
# compute eigengene-based connectivity (kME):
seurat_obj <- ModuleConnectivity(
seurat_obj,
group.by = 'cell_type', group_name = 'INH'
)重命名
# rename the modules
seurat_obj <- ResetModuleNames(
seurat_obj,
new_name = "INH-M"
)我们可以使用该函数可视化按 kME 排序的每个模块中的基因
# plot genes ranked by kME for each module
p <- PlotKMEs(seurat_obj, ncol=5)
p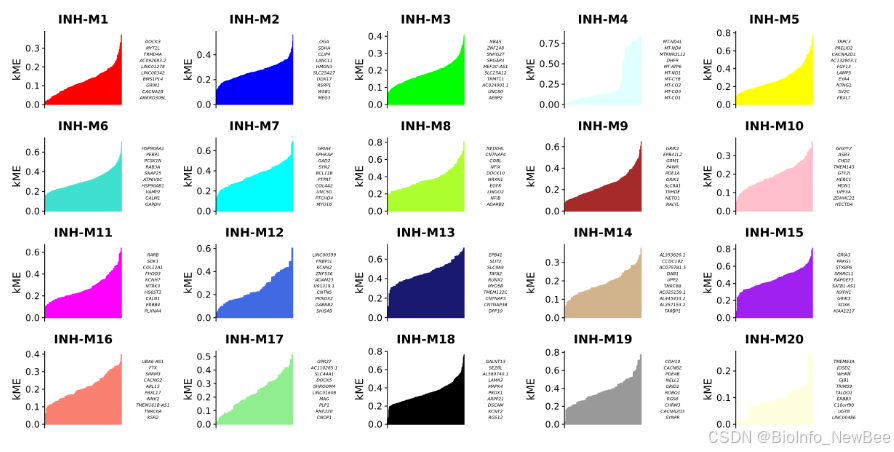
获取模块分配表
hdWGCNA 允许使用该函数轻松访问模块分配表。该表由三列组成:存储基因的符号或 ID,存储基因的模块分配,并为每个模块存储颜色映射,这在许多下游绘图步骤中使用。
# get the module assignment table:
modules <- GetModules(seurat_obj) %>% subset(module != 'grey')
# show the first 6 columns:
head(modules[,1:6])可以使用该函数提取按 kME 排序的前 N 个枢纽基因表。
# get hub genes
hub_df <- GetHubGenes(seurat_obj, n_hubs = 10)
head(hub_df)保存
saveRDS(seurat_obj, file='hdWGCNA_object.rds')
















 3267
3267

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









