




 本文详细介绍了微生物菌群多样性分析的实验流程,包括样本的OTU划分、Alpha和Beta多样性分析,以及多样性的各类指数计算。通过序列预处理、质量控制,使用QIIME等工具进行序列比对和分类,分析丰度分布和样本间差异,揭示菌群结构和变化。此外,还涵盖了系统发育树构建、可视化方法,如GraPhlAN和热图分析。
本文详细介绍了微生物菌群多样性分析的实验流程,包括样本的OTU划分、Alpha和Beta多样性分析,以及多样性的各类指数计算。通过序列预处理、质量控制,使用QIIME等工具进行序列比对和分类,分析丰度分布和样本间差异,揭示菌群结构和变化。此外,还涵盖了系统发育树构建、可视化方法,如GraPhlAN和热图分析。
参考链接https://www.docin.com/p-2107733531.html
在开始实验项目之前,明确实验流程,一步一步获取实验结果,以可视化工具展现结果,并加以生物学意义上的分析,获取完整的分析报告。下面给出菌群多样性分析报告中应包含的大致内容,并对实验流程做出指导性的阐述。
1.项目背景与概况
简单阐述所研究项目的国内外现状,本项目的目的与意义。
2.材料与方法(material and methods)
获取材料的方式主要有:现有数据库下载生物序列;测序实验采集;方法,这一部分内容主要阐述实验流程,各个步骤所采用的技术、算法、工具。
2.1原始数据测定实验流程

2.2数据分析流程
利用原始测序数据以及样本的分组信息进行一系列数据分析,流程如下。
(1)序列预处理和质量控制
(2)OTU划分与分类
划分得到样本的OTUs.获取各OTU的代表序列,通过各OTU所包含的reads数目,创建OTU_table;用于分类level鉴定和系统发育学分析。该分析过程可用一些可视化工具进行分析描述.
Q:利用什么方法划分样本的OTU?
各种工具,如:QIIMEE软件调用UCLUST序列比对工具,按照97%的序列相似度进行归并和OTU划分
Q:如何选取OTU代表序列?
每个OTU中丰度最高的序列为该OTU的代表序列
利用已获取的代表序列进行界门纲目科属种的分类,将代表序列和对应数据库的模板序列相比对,获取每个OTU所对应的分类学信息,常用的模板序列数据库有:GreenGenes,RDP,Silva
Q:划分后的OTU丰度不同,丰度极低和丰度极高的OTU出现频率低,丰度极低的OTU非常稀有,相当于菌群数据中的噪音,应该进行去除。
共有OTU通过VENN图来展示。
(3.1)样本的Alpha多样性分析
Rarefaction稀疏曲线分析,通过稀疏曲线判定测序深度是否达标。在各样本中随机抽取reads进行OTU划分,OTU数目稳定在一个数值范围内,则表明测序深度达标。
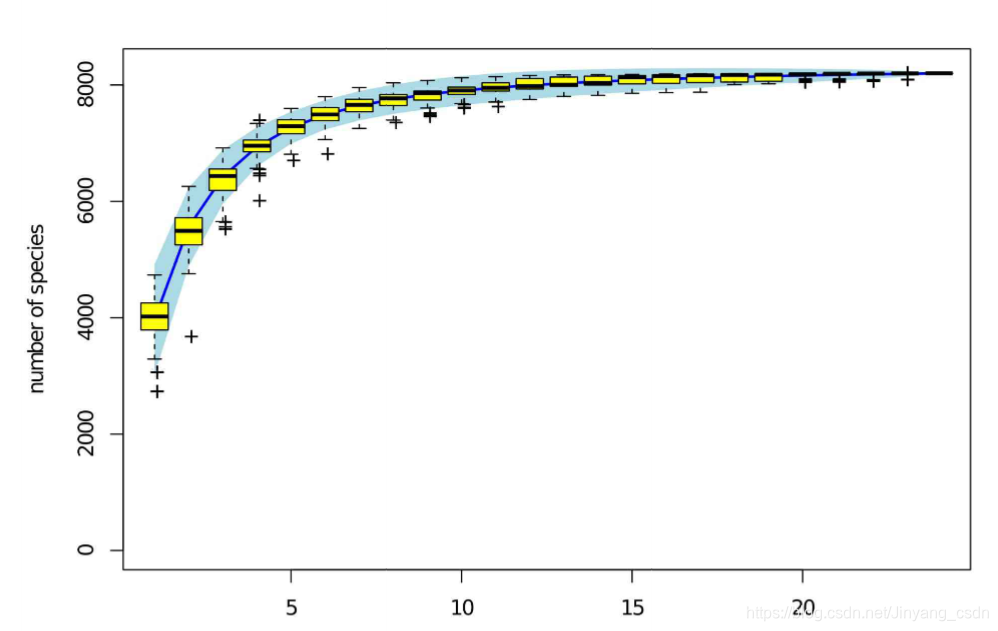
Specaccum物种累积曲线分析,不断增加样本数目,进行OTU划分,OTU数据稳定,则达标;随机抽取不同的固定数目的samples,划分OTU,得到OTU 数目的均值,极大小值,上下边缘,绘制盒图,按照一定规律(递增)改变samples数目,再多次抽样,制作OTU盒图。该图像能描述样本采样的充分性与物种丰富度。
Abundance Range丰度等级曲线分析,将样本按照丰度从大至小排序,展现丰度在样本之间的差异。
多样性指数计算,体现群落丰富度的丰度指数(Chao1和ACE),多样性指数(shannon和simpson)
(3.2)Beta多样性分析
有基于序列的Beta分析(欧氏距离,bray curtis距离等),基于系统进化的Bata分析 (根据系统发生树进行比较)
PCA/PCoA/NMDS排序分析
PCoA与PCA的区别在于PCA是基于原始的物种组成矩阵所做的分析,使用的是欧式距离,仅仅比较的是物种丰度的不同,而PCoA首先根据不同的距离算法计算样品之间的距离,然后对距离矩阵进行处理,使图中点间的距离正好等于原来的差异数据,实现定性数据的定量转换。百分比含义同PCA。
非度量多维标定法(Non-MetricMulti-Dimensional Scaling,NMDS)是一种适用于生态学研究的排序方法,主要是将多维空间的研究对象(样本或变量)简化到低维空间进行定位、分析和归类,同时又保留对象间原始关系的数据分析方法。类似于 PCA 或者 PCoA,通过样本的分布可以看出组间或组内差异。NMDS原设计的目的是为了克服以前排序方法中包括PCA、PCoA在内的缺点,即线性模型。NMDS的模型是非线性的,能更好地反映生态学数据的非线性结构,有的研究认为NMDS的效果优于PCA/PCoA
UPGMA聚类分析
基于以上方法分析得到的距离矩阵,可用UPGMA(Unweighted Pair-group Method with Arithmetic Mean),分析样品间的差异。非加权组平均法,是一种常用的聚类分析方法。其原理是:假定的前条件是在进化过程中,每一世系发生趋异的次数相同,即核苷酸或氨基酸的替换速率是均等且恒定的。通过 UPGMA 法所产生的系统发生树,通过树枝的距离和聚类的远近可以观察样品间相似性
多组UniFrac距离比较
三元相图分析
(3.2.1)菌群比较分析和关键物种筛选
略,随着研究深入再展开
CAP分析
RDA分析
随机森林分析
(3.3)分类组成分析
各分类level(界门纲目科属种)的微生物类群统计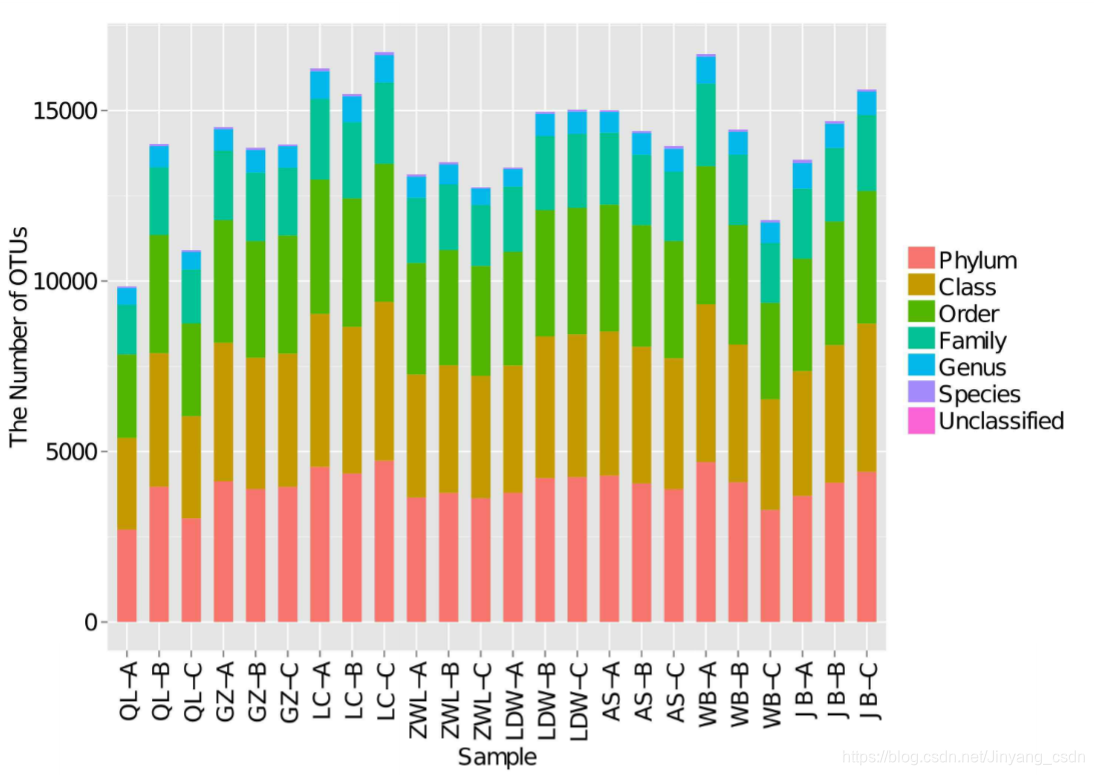
各分类level的丰度分析
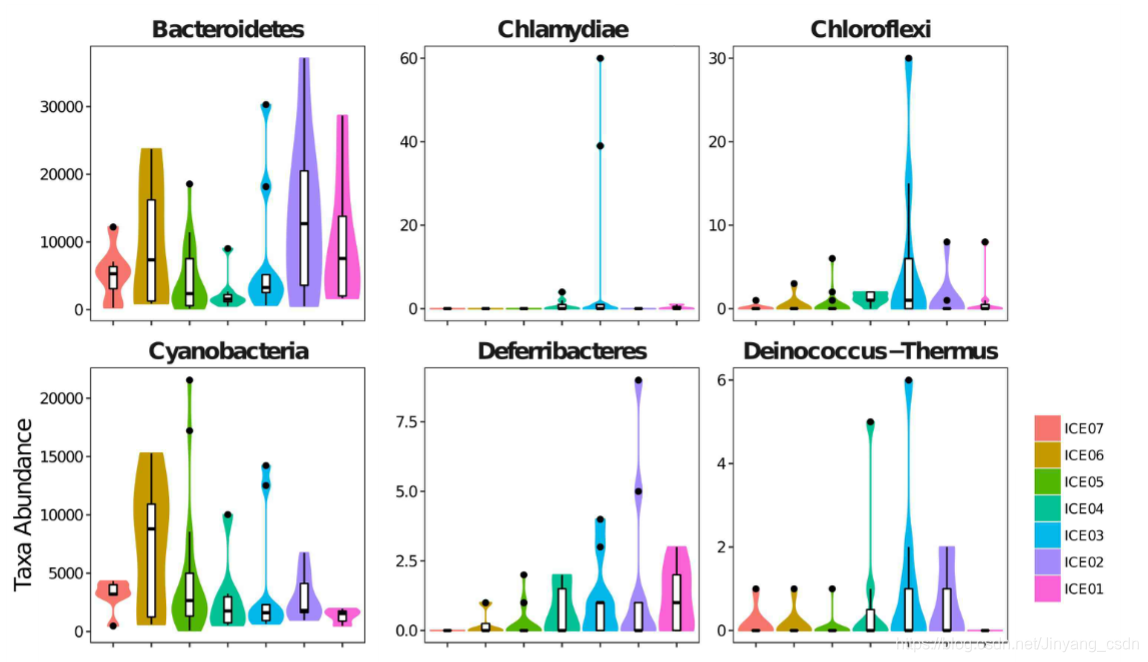
纵坐标为各个OTU的相对丰度,越长表示相对丰度越高
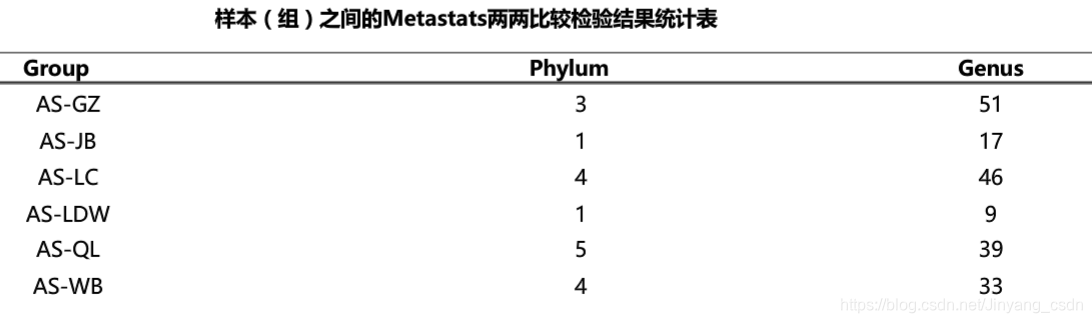
Metastats组间差异分析
利用Mothur软件,条用Metastats,对门和属水平的分类level在两两样本之间的序列量差异进行比较。
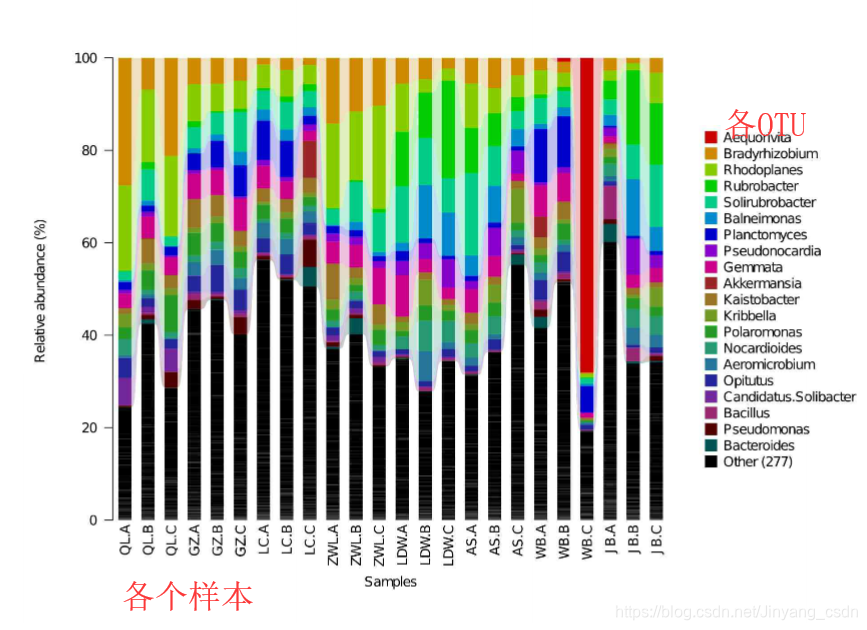
可通过图形进一步呈现各OTU在样本内的丰度分布
LEfSe组间差异分析
基于LDA的方法,目标是筛选关键的Biomarker,提交属水平的相对丰度矩阵到galaxy在线平台,可自动分析,并可视化。
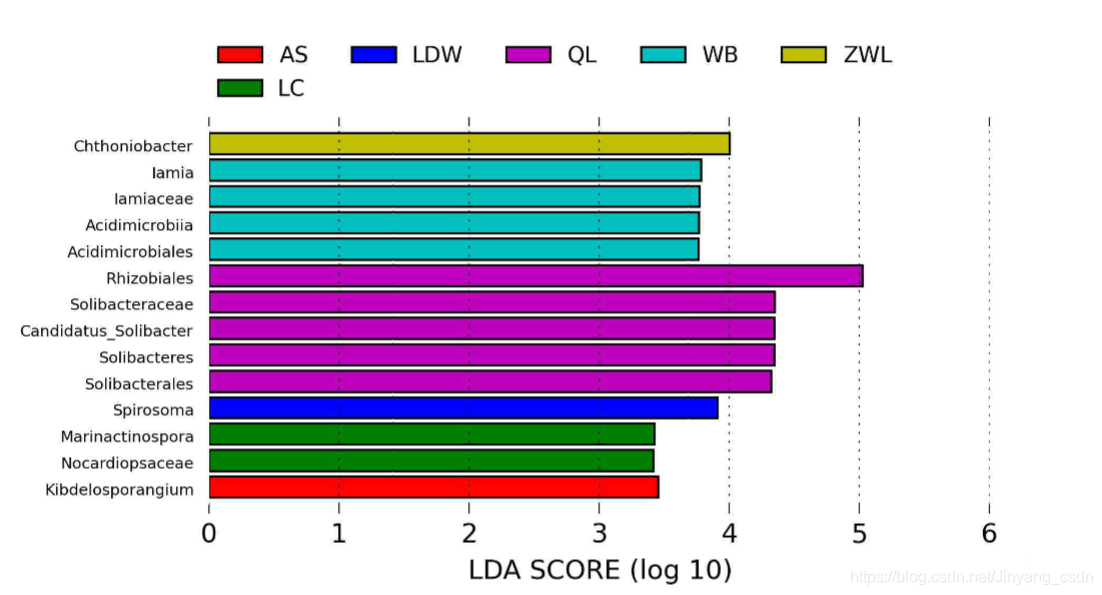
长度越长代表其特征越明显,越适合做样本的biomarker
(3.4)菌群组成交互式可视化
系统发育树构建
GraPhlAN可视化展示
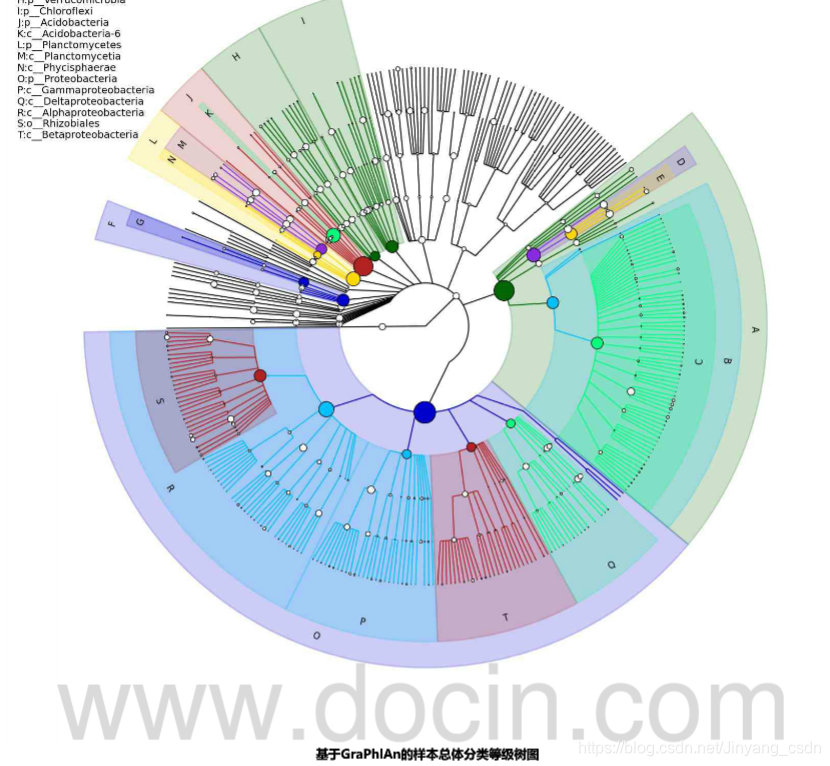
从门到属的所有OTU的等级关系,节点大小表示平均相对丰度
优势物种/OTU热图分析
(3.4.1)网络分析
略,随着研究深入再展开

















 2529
2529

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









