



RMSD,Root Mean Square Deviation,均方根偏差;RMSF,Root Mean Square Fluctuation,均方根波动。
在轨迹分析中,最经常用,最简单,也最有用的就是这两巨头,二者都是对位移的平方和再求平方根,最后求得均值。其实差别就在于,这个“均值”是哪个物理量按照什么的平均。
举例子,现在跑了1ns的轨迹,2fs保存一帧,一共50w帧。假设以初始结构为参考构象,则RMSD曲线就是需要遍历计算50w帧结构与参考构象的RMSD值(当然你也可以减小帧数),然后将每一帧的RMSD连起来的结果。其中,轨迹中某一帧相对于参考构象的RMSD的计算方法如下:
计算RMSD的时候,公式如下:
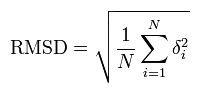
δi就是某一帧的第i个原子的位置减去参考构象中它的位置(位置偏移量),然后取所有原子的偏移量的平方和,然后对原子数N取平均,然后开方,就是这一帧结构相对于参考构象的RMSD。
同样是这个轨迹,现在我要求RMSF,公式如下:
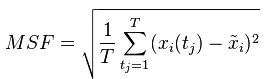
可以看出来,里面引入了T(时间)这个变量,说明和时间有关。(Xi(tj)-xi)就是t时刻某个原子的位置减去初始时刻它的位置(也是位置偏移量),然后取所有时刻(可以理解为50w个2fs)的偏移量的平方和,然后对时间T取平均,然后开方,就是这个原子在时间T内,相对于初始时刻的RMSF。
到这里,区别就明显了:
RMSD一般是说的某个时刻相对于参考构象的结构偏差;
而RMSF是说的一段时间内,某一个原子相对于参考构象的结构变化,反应了原子的自由度(灵活性)

















 4279
4279

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









