




注意!文章中的代码有问题,注意修改
1.
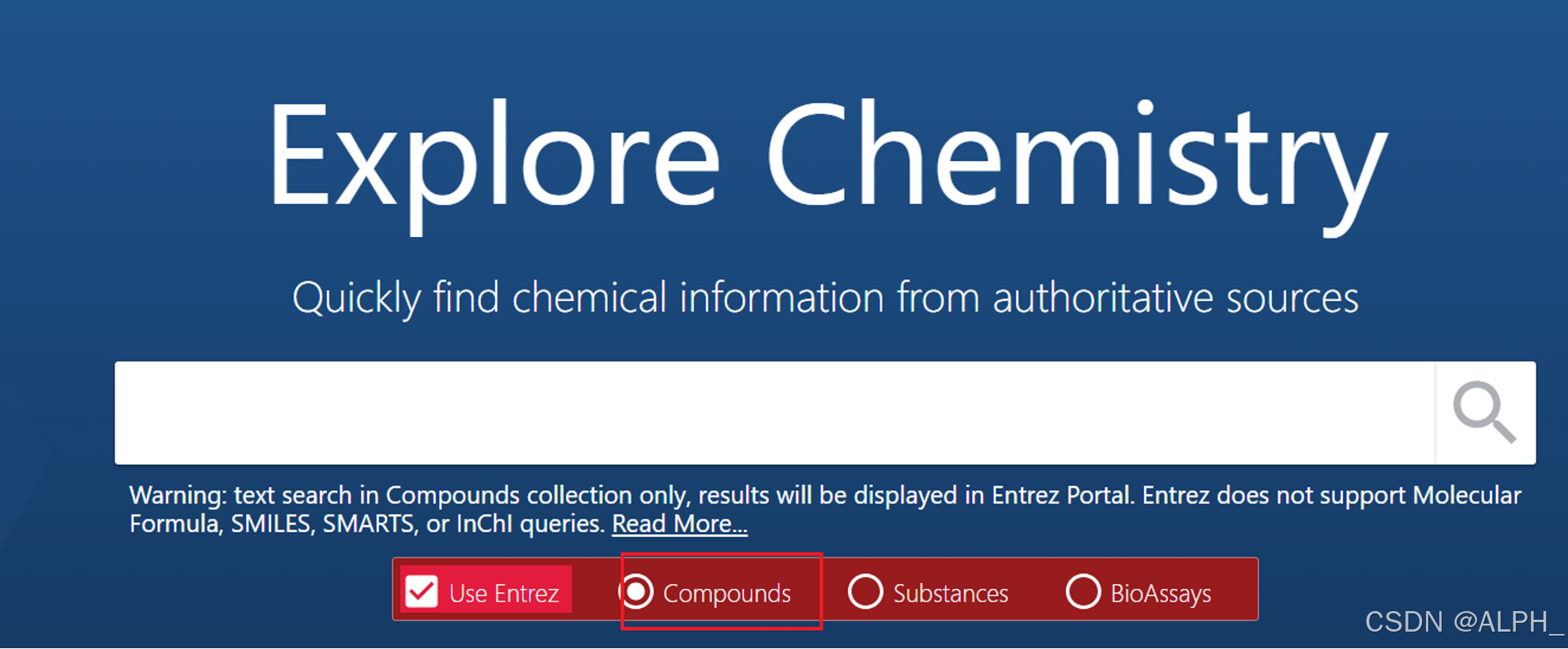
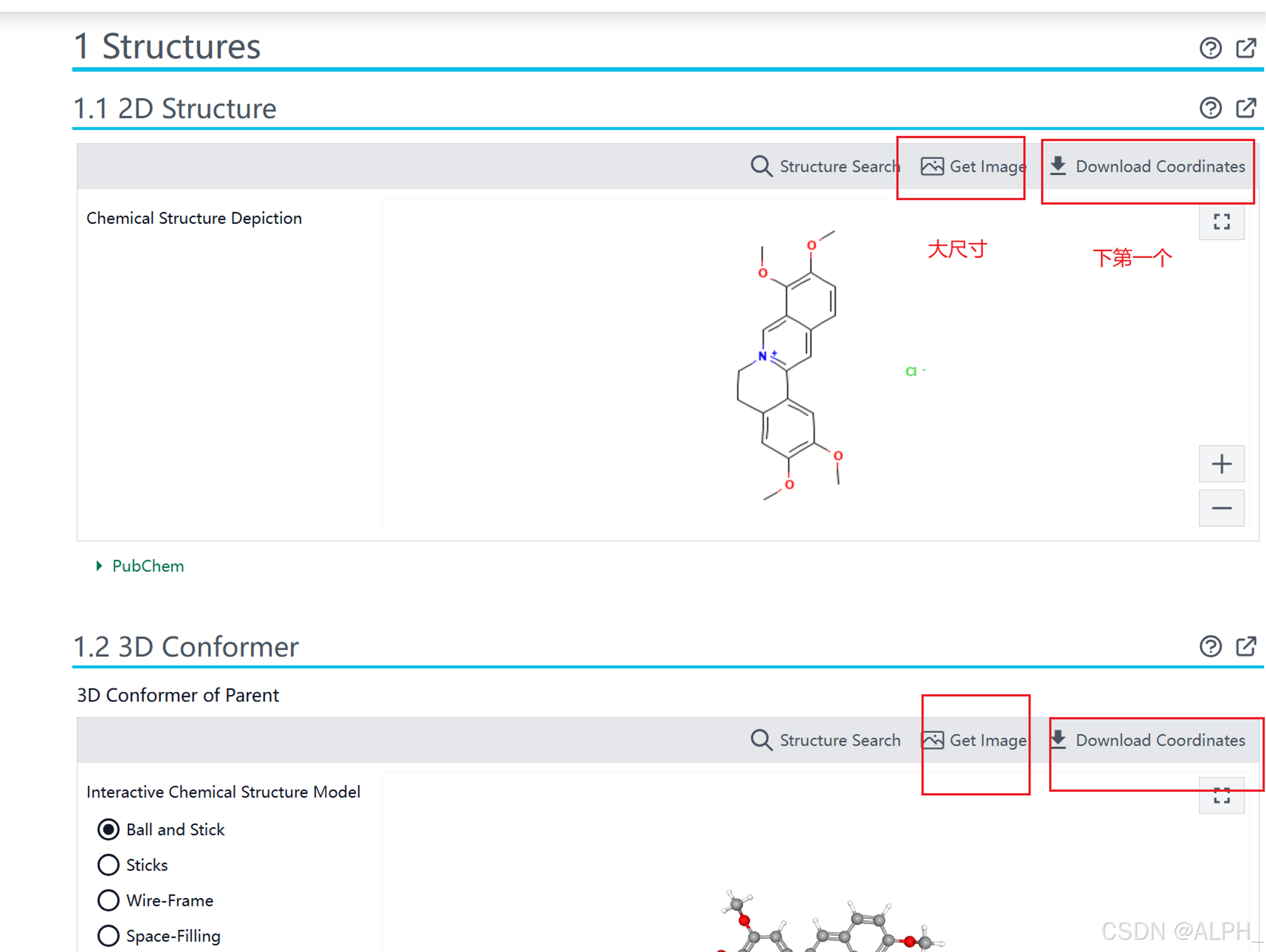
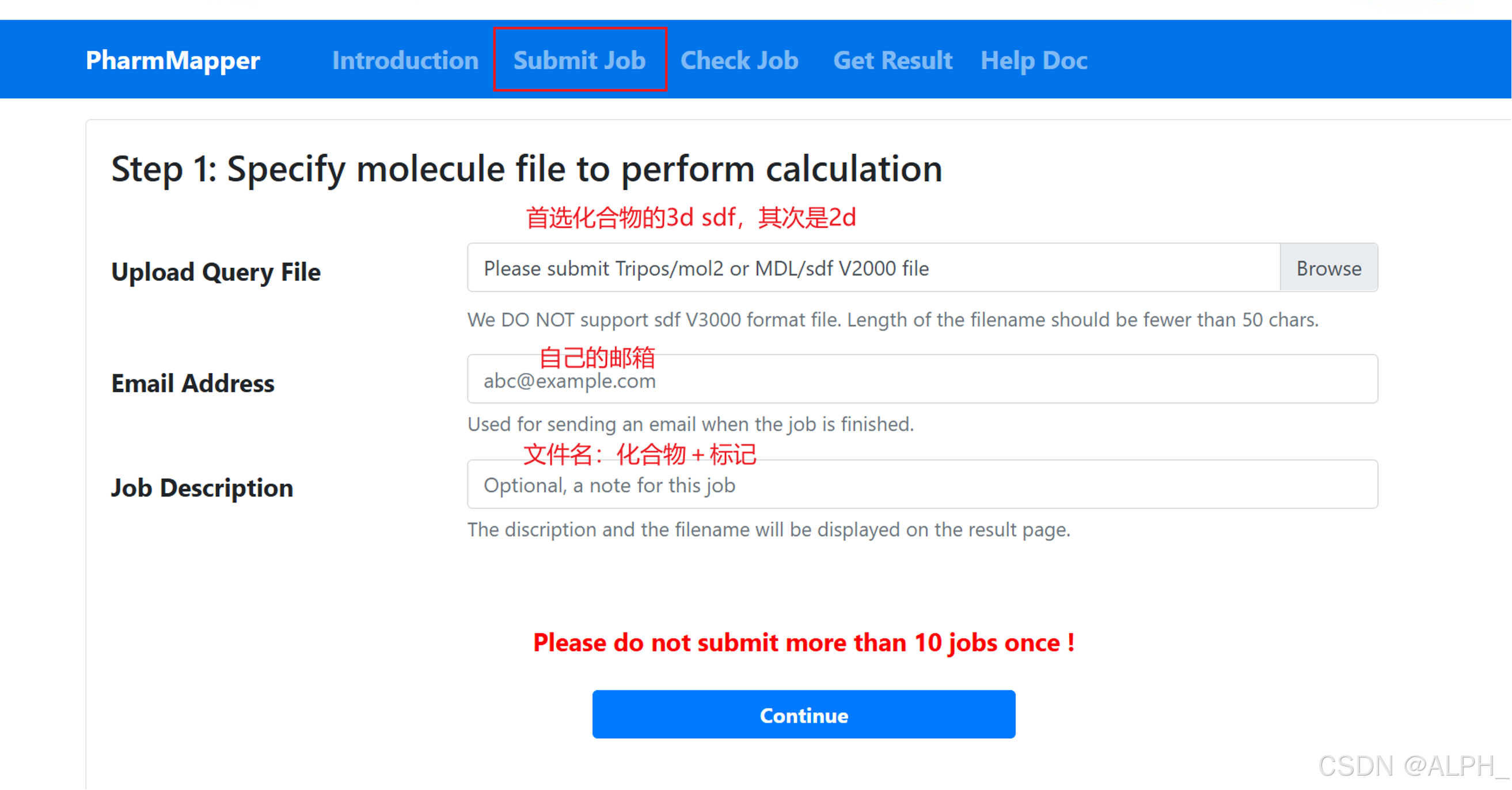
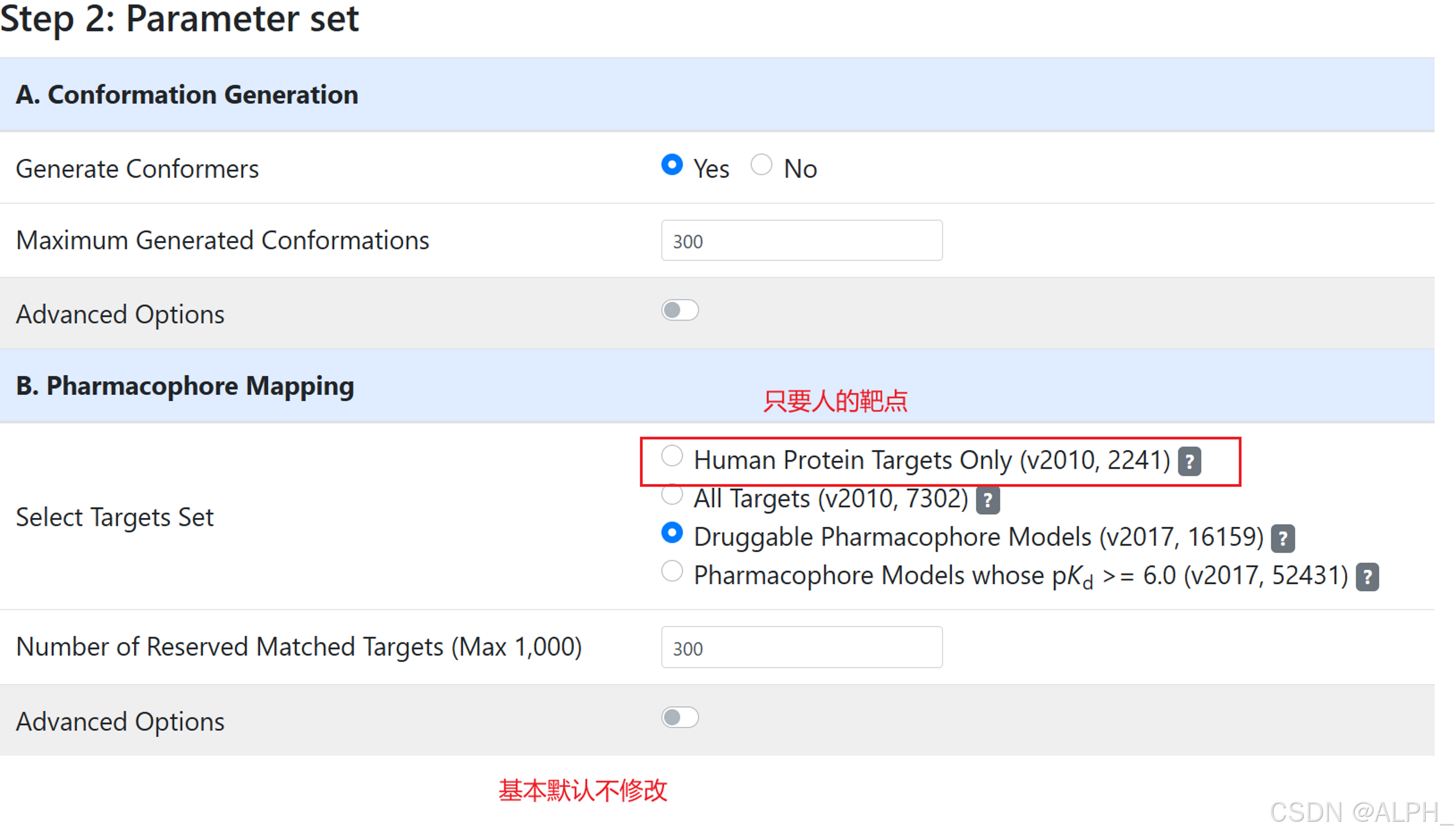
保存此时的网页,等待结果,或者查看邮箱(时间是几个小时或几天)
然后把结果保存

图形可以放大保存
文件结果

添加靶点基因名称信息便于后续分析。
1.注释文件包含基因名称和数据库ID的对应关系。
2.注释文件第二列为基因名称,第一列为基因ID。
3.注释文件由多个数据库的ID转化为基因名字,下载并整合多个数据库的信息。
4.做人的话直接用这个注释文件就好。其他物种要买一下
<<ann.txt>>
针对这三个文件进行操作
就是从excel复制出来,然后新建一个input.txt,把我们这个文件拷贝到这个input.txt就完成了。

在含有perl的文件夹里运行程序,获得out文件复制到excel里
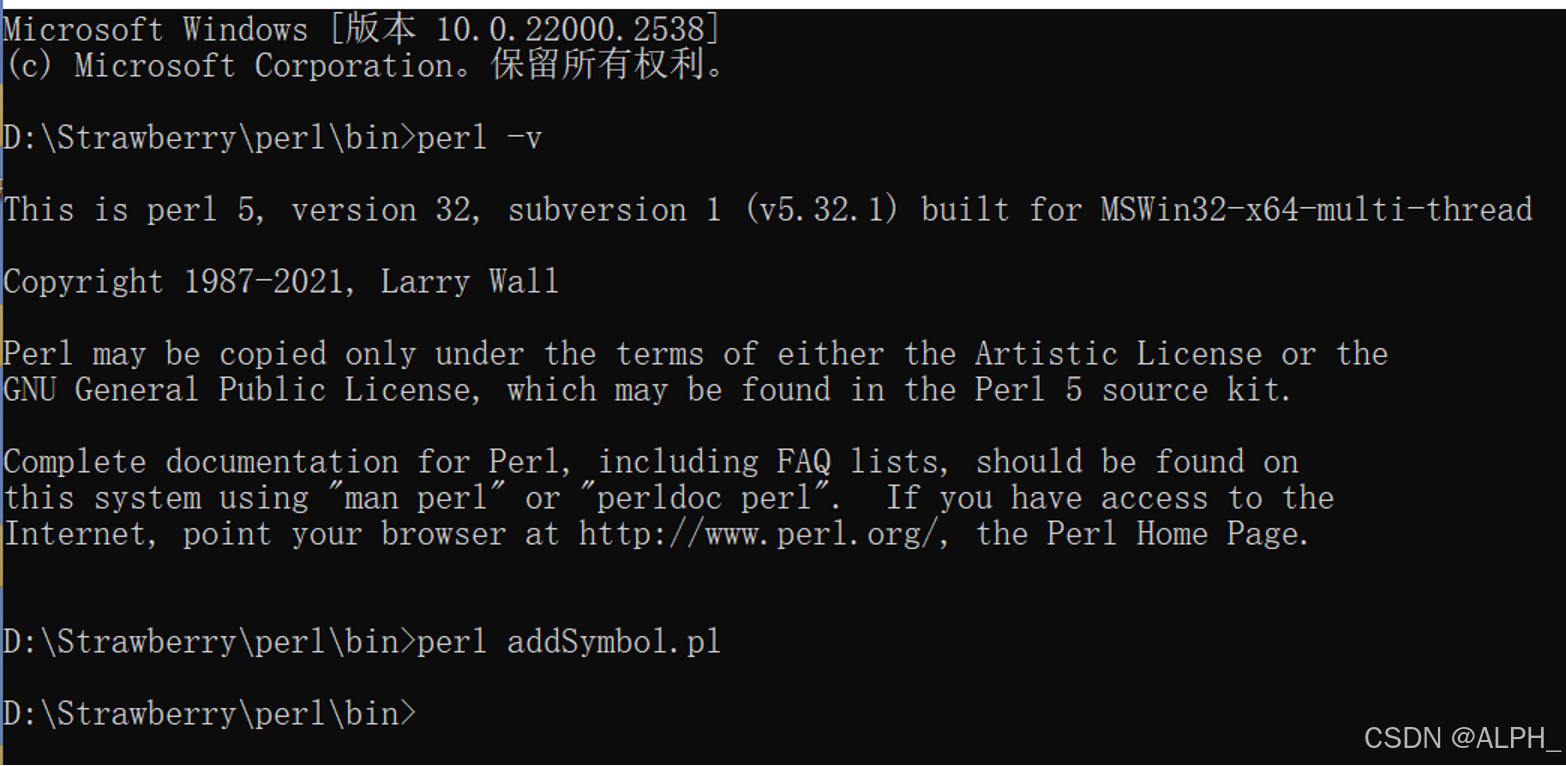
通常会留下这几行文件

提取其中两行形成merge.xlsl→一列是靶点另一列是化合物名称
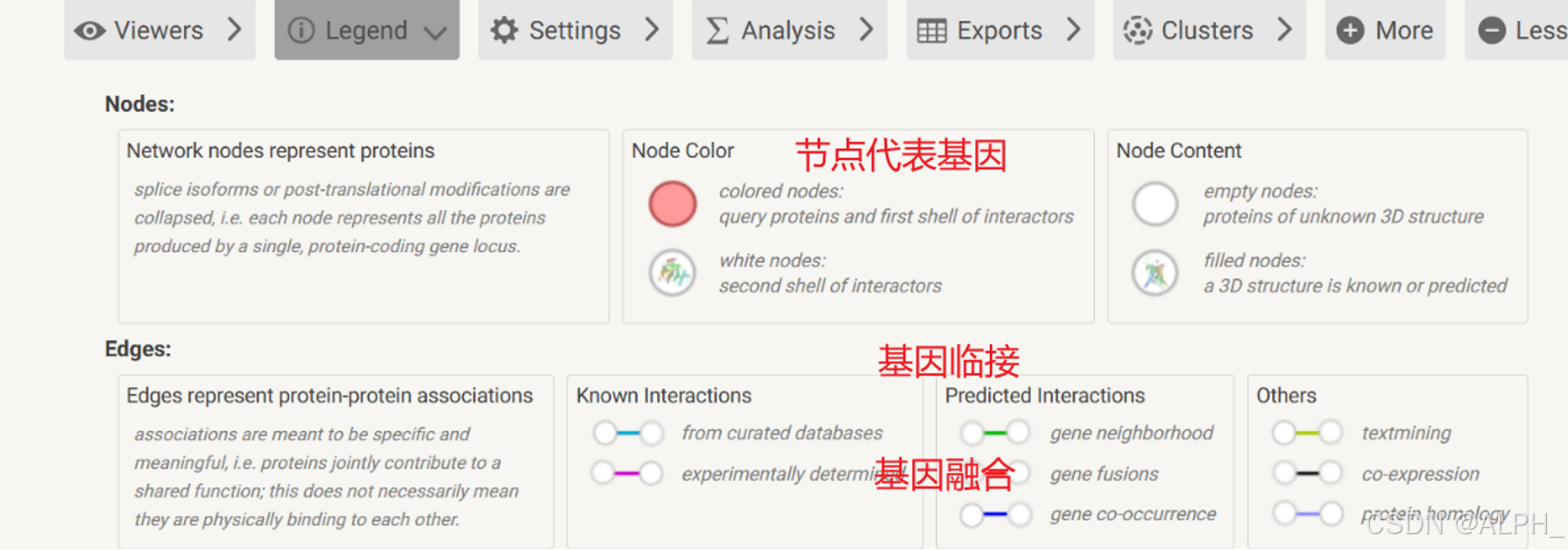
做蛋白互作网络:STRING: functional protein association networks
基因名字:基因名字不要太多了,如果太多,可以选择分值进行过滤,也可以每个化合物选择前50个基因进行一个分析。
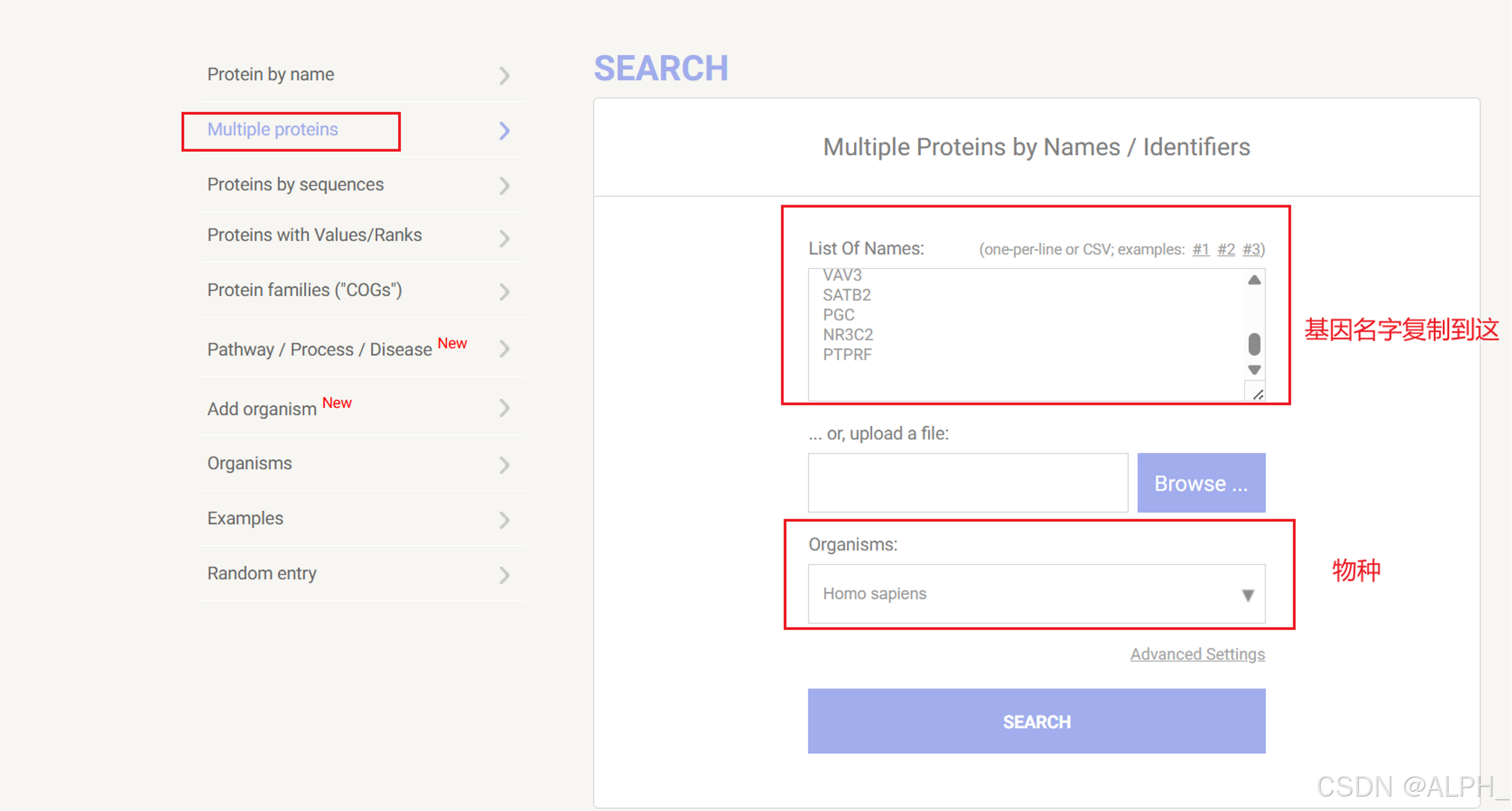
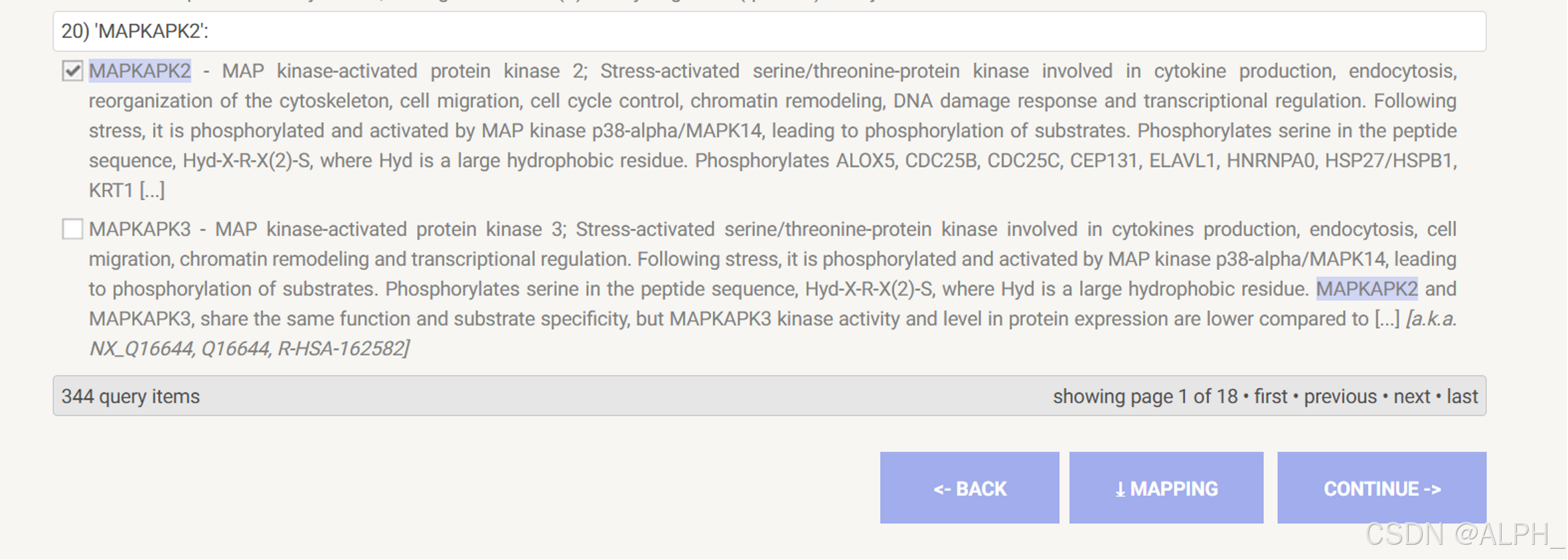
拉到最后点continue(确保全搜索完)
一般默认
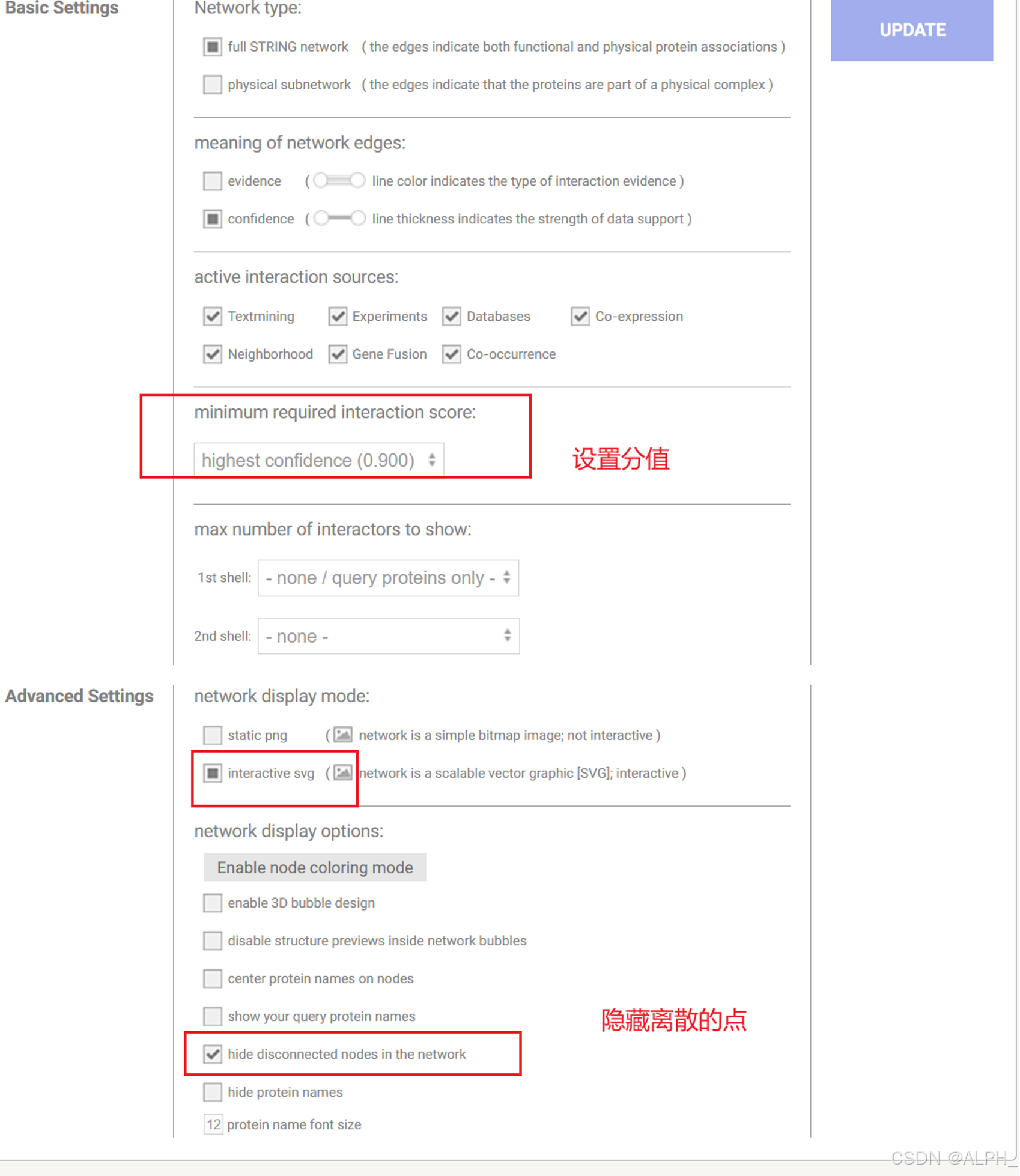
分值就是过滤的分值,它的分是0到1分值越大就是两个基因之间具有蛋白相互作用的可能性越大。
如果发现还是很乱,就把数值调到0.96
第二部分选evidence
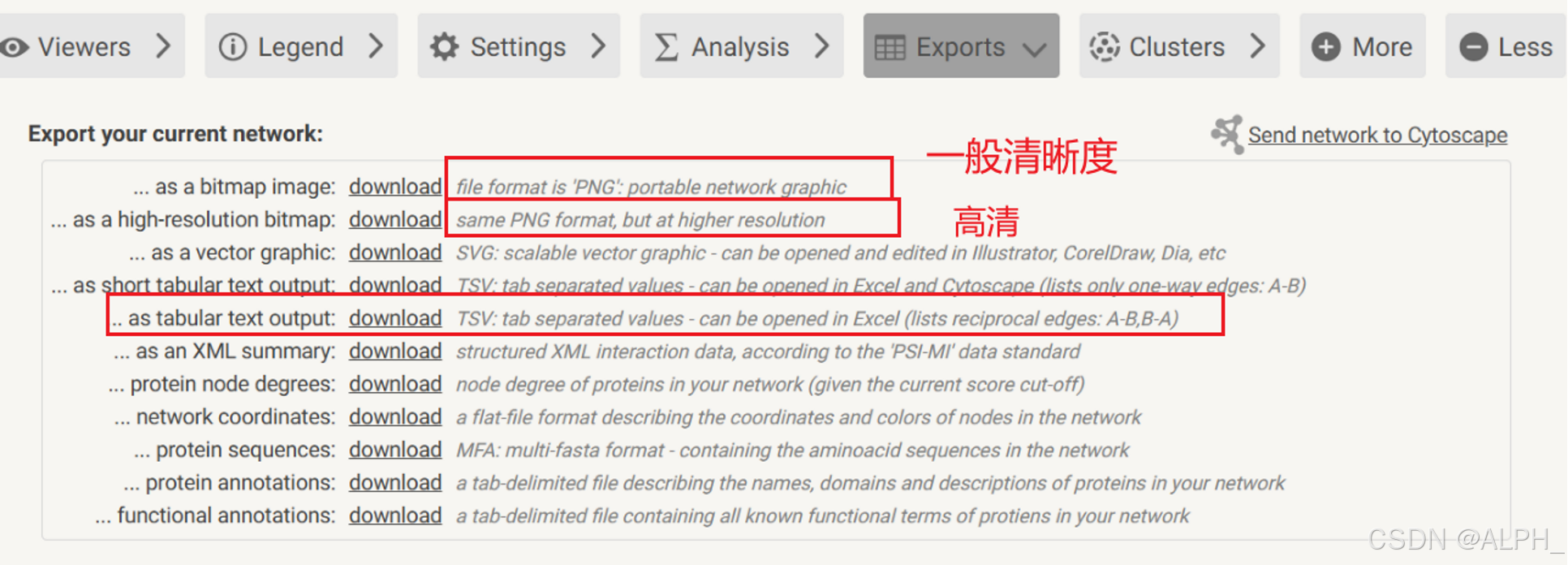
需要下载的格式

把merge.xlsl复制成merge.txt
准备好这三个文件
获得这三个文件
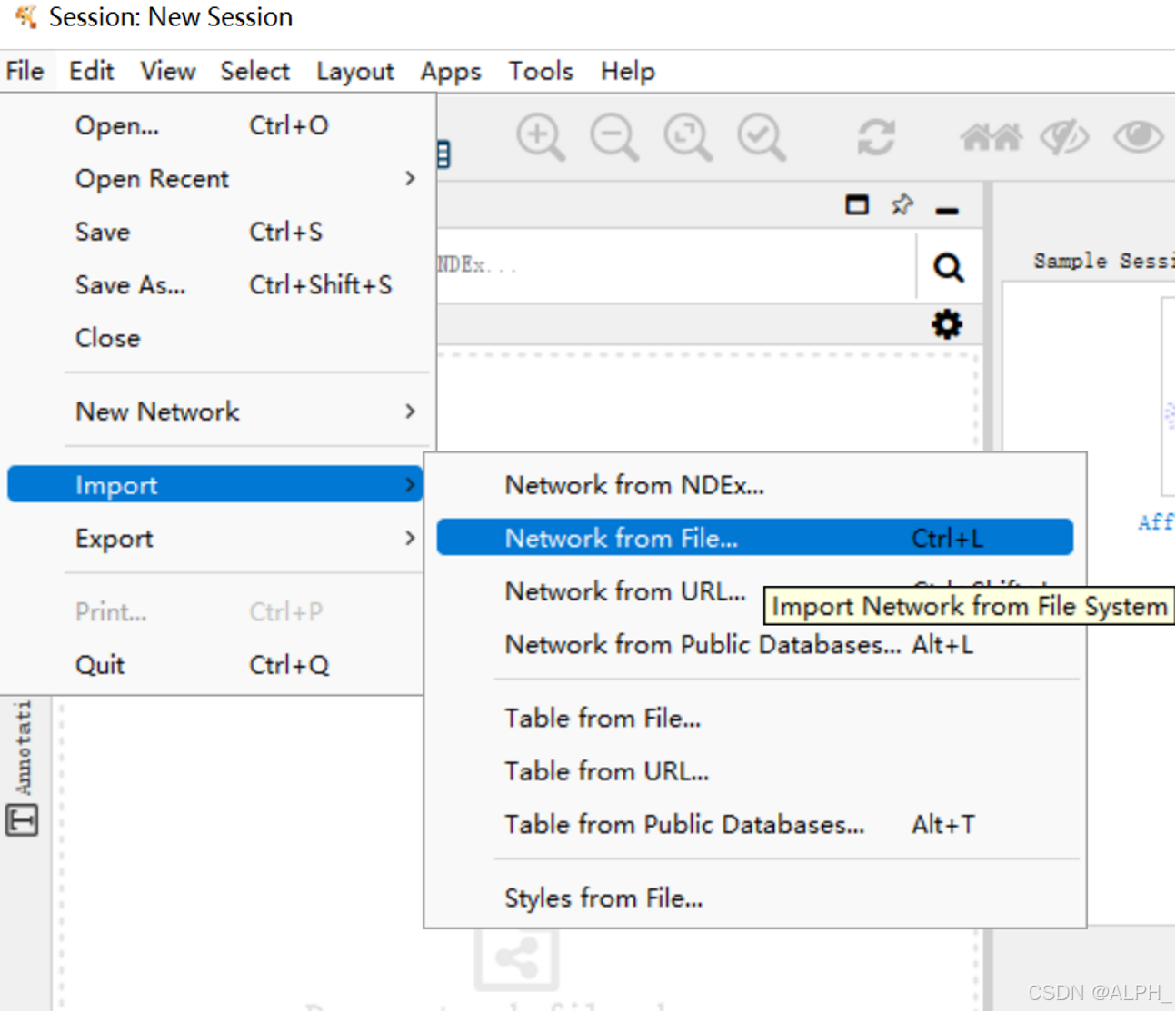
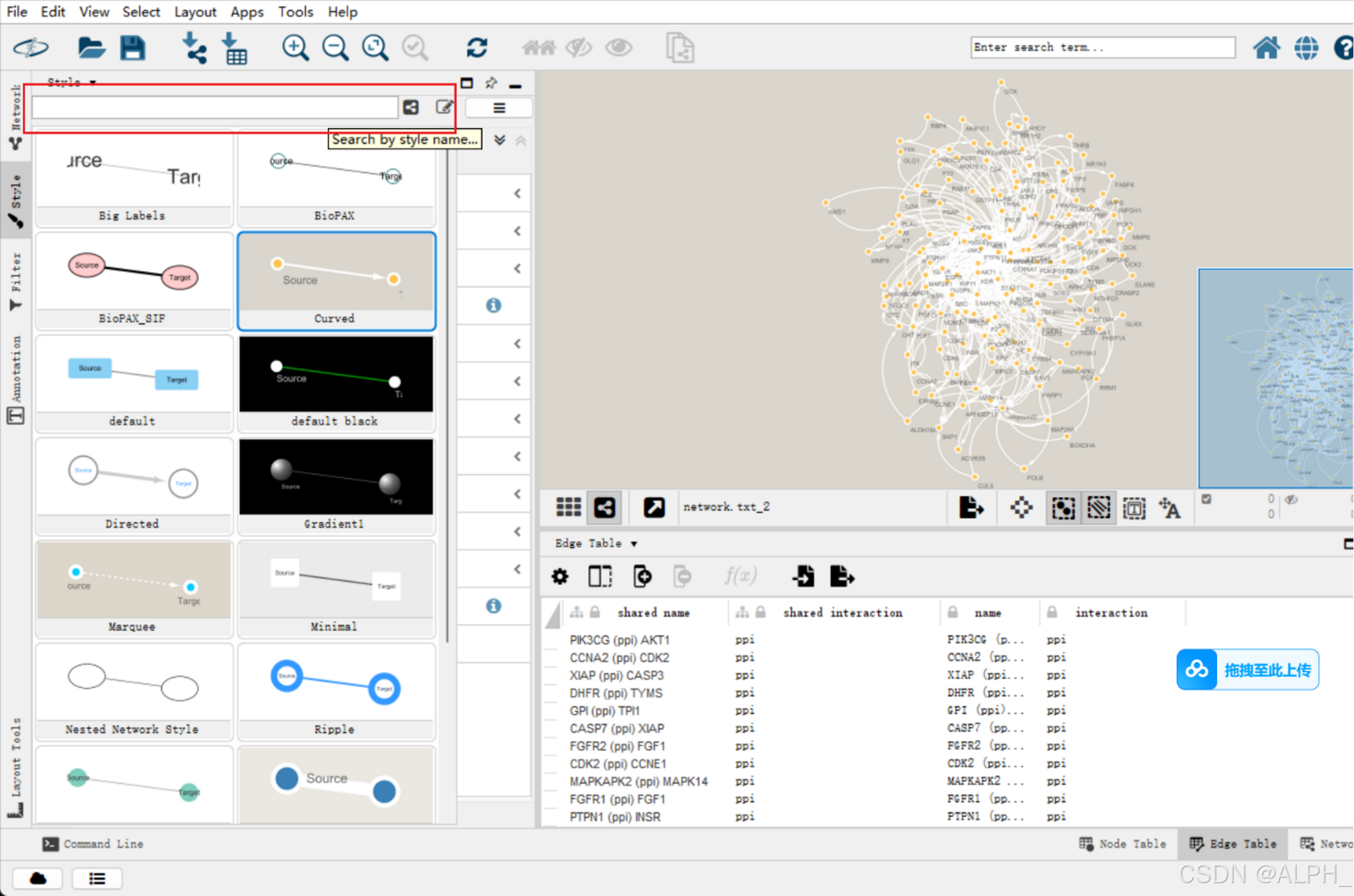
导入network.txt
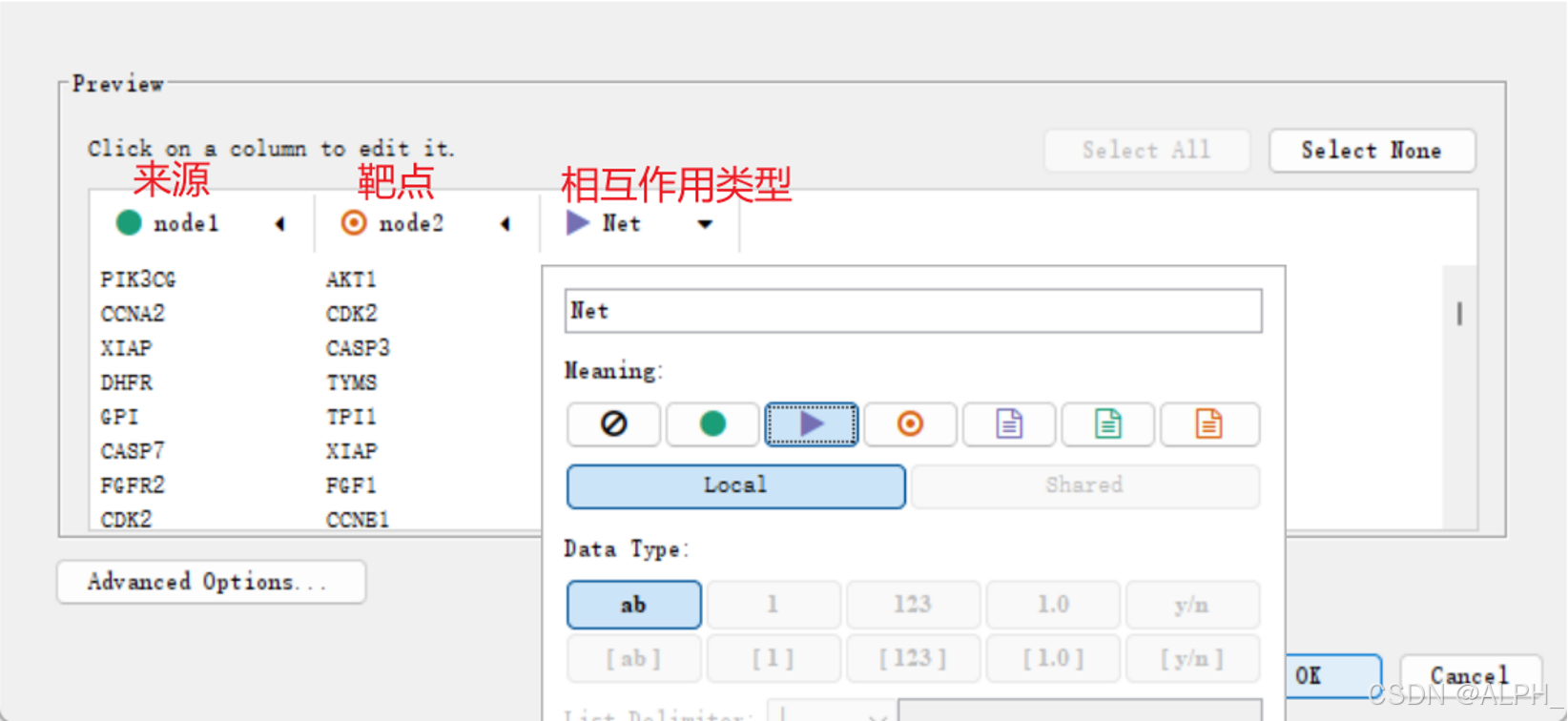
选择类型
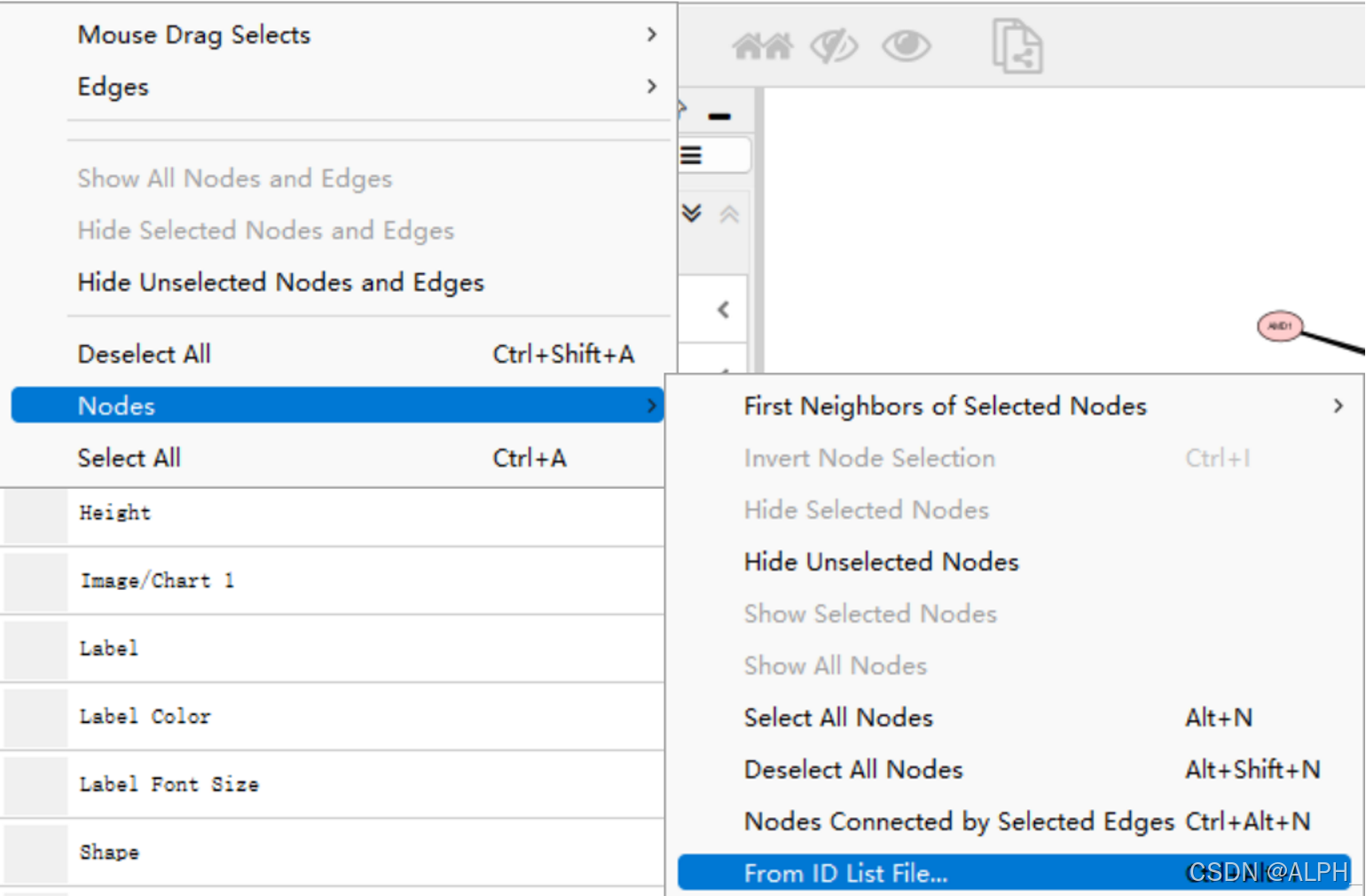
Select geneList.txt
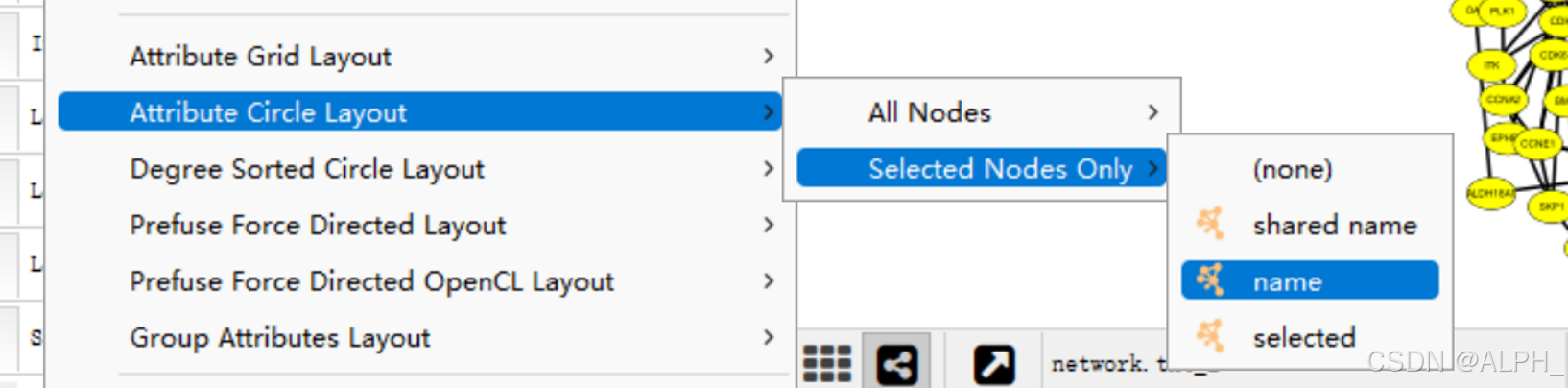
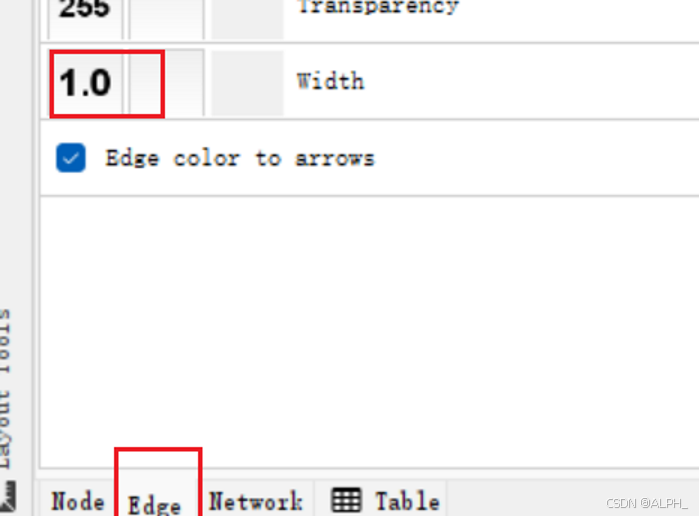
调整线条
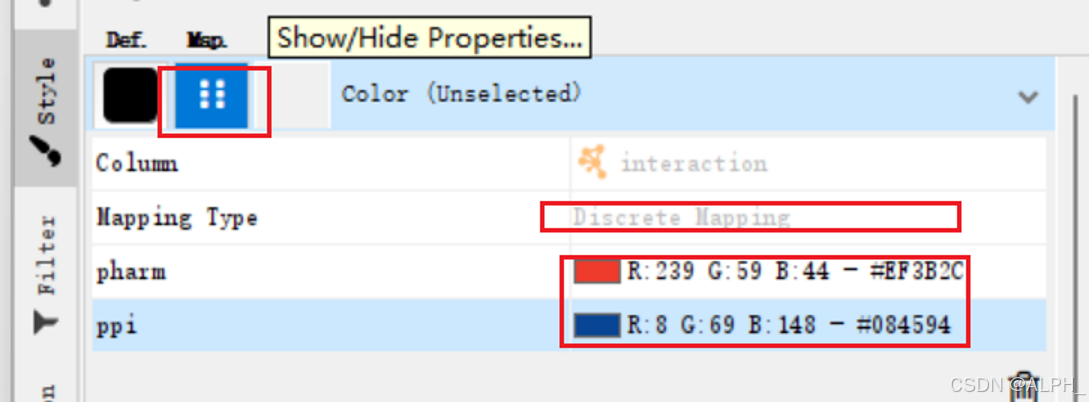
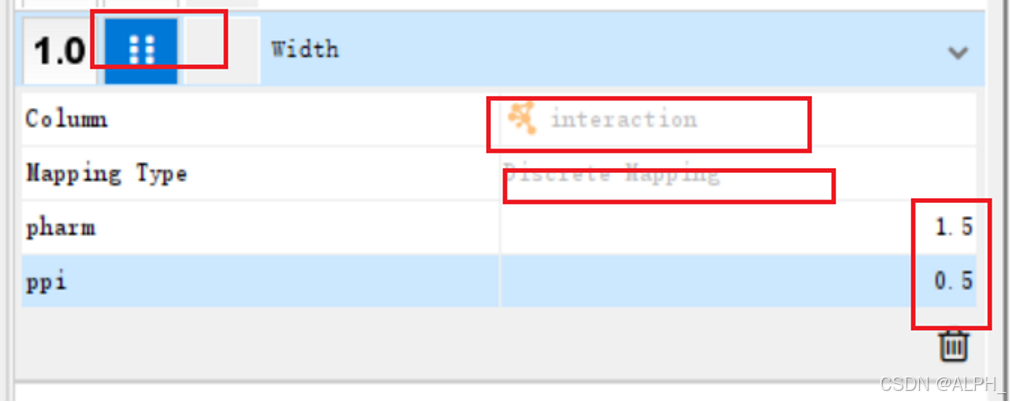
现在node的属性还没导入(node.txt)
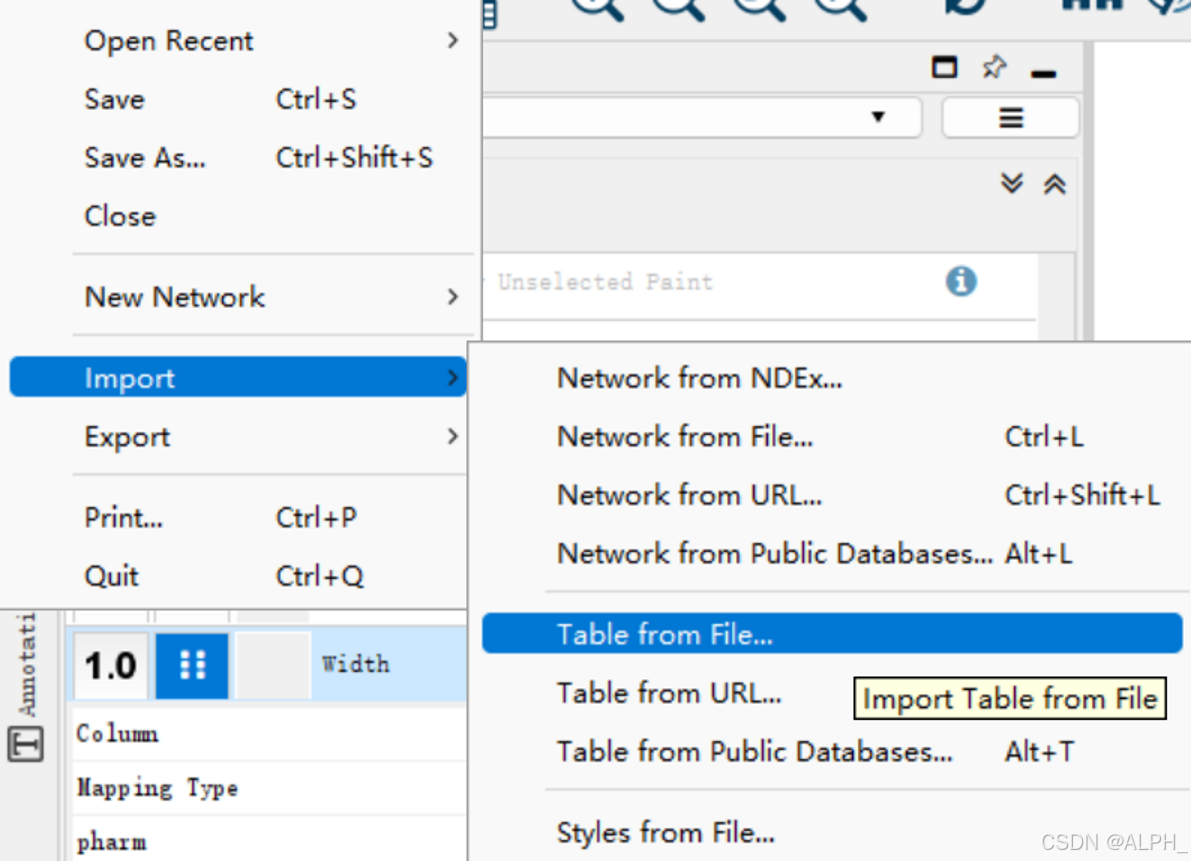
总的来说就是按照自己的需求去个性化(这里不多赘述了)
保存图形:保存时注意,只会保存方框里面的东西
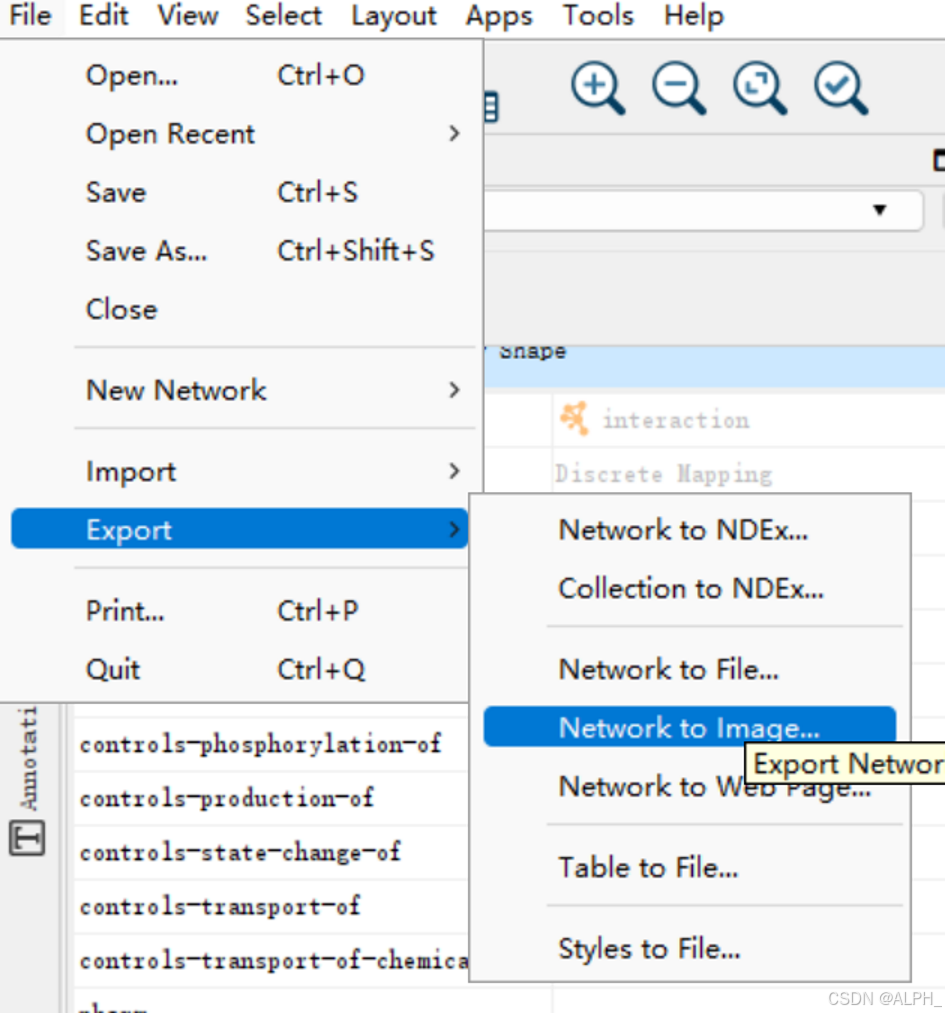
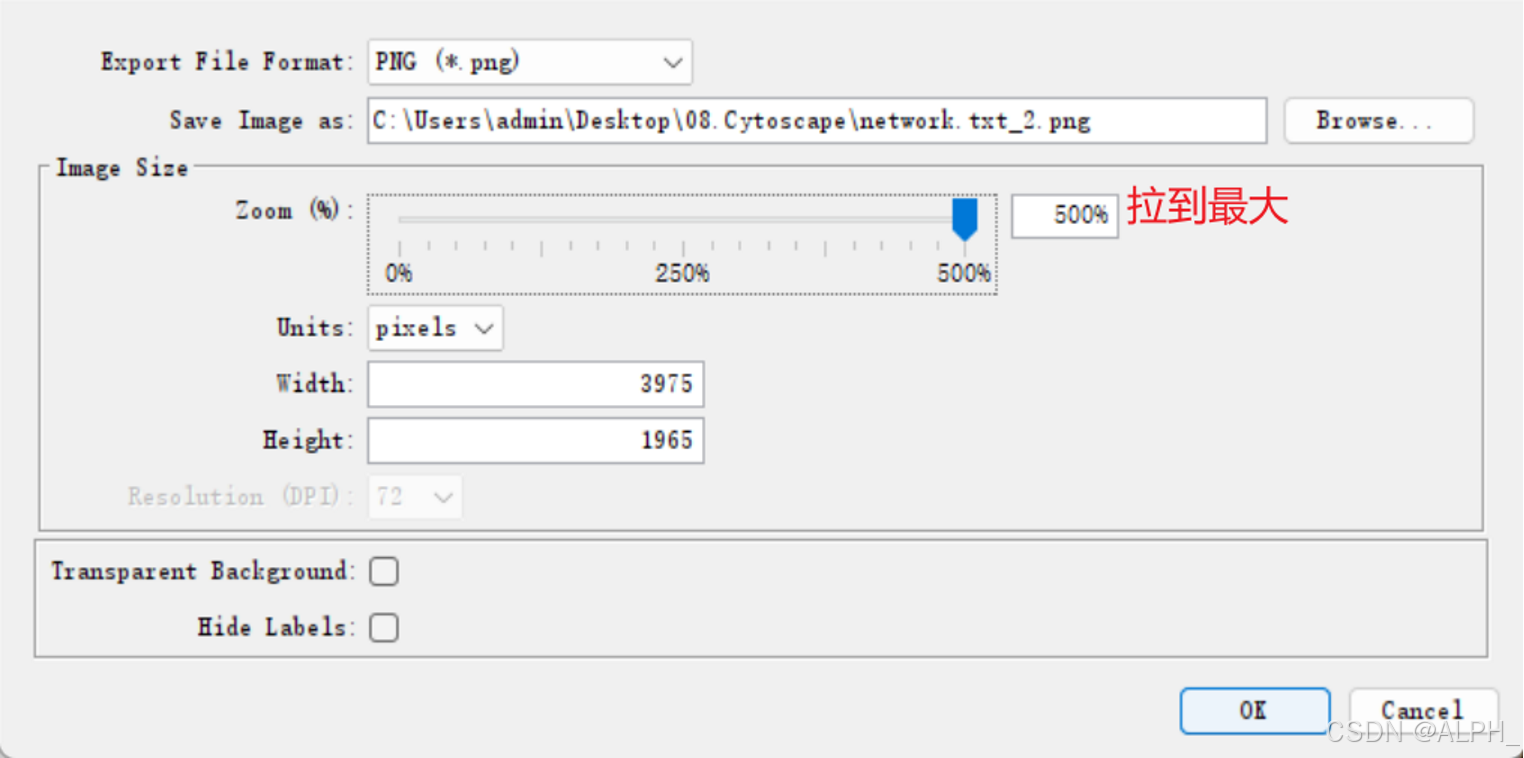
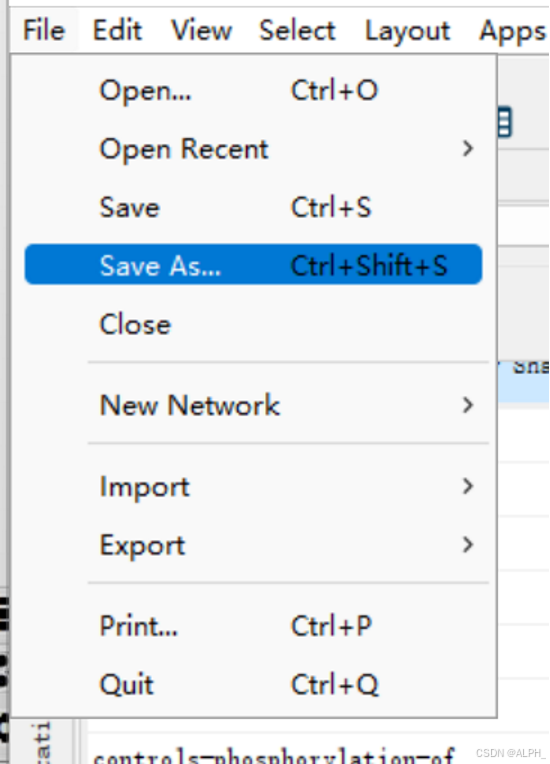
保存网络:

















 3552
3552



 到【灌水乐园】发言
到【灌水乐园】发言








