



工具链接:https://www.henbio.com/tooldetail?id=314
在基因组学研究领域,准确地揭示基因组中哪些区域是开放的并且处于活跃状态,对于理解基因的表达调控机制至关重要。目前,ATAC-seq(测序抗体转座酶可及性染色质分析技术)是实现这一目标的关键技术之一。ATAC-seq技术的核心优势在于其对细胞样本的需求量极低,这使得它能够在样本稀缺的情况下依然能够高效地开展研究工作。凭借这一独特的优势,ATAC-seq已经被广泛应用于多个重要的研究领域,包括但不限于染色质结构的解析、表观遗传学的深入研究、转录因子结合位点的精准定位以及基因调控网络的全面解析等。为了进一步降低基因组学研究的门槛,让更多的研究人员能够便捷地利用ATAC-seq技术开展研究工作,HiOmics平台推出了ATAC-Seq染色质可及性分析工具,为研究人员提供了强大的支持。特别是对于那些刚刚踏入这一领域的新手来说,HiOmics平台的友好性体现在其零代码在线运行分析的功能上。这意味着,即使是没有编程基础的研究人员,也能够轻松地上传自己的数据,通过平台的图形化界面进行操作,快速启动分析流程,并在短时间内获得可靠的分析结果。这种高效且易于上手的分析方式,无疑为推动基因组学研究的普及和发展提供了有力的助力。
下面我们看一下ATAC-Seq分析具体操作。
1.首先点击进入ATAC-Seq分析。
界面右侧对分析工具的使用方法及结果解读做了详细说明。左侧是上传的样本数据文件选项,对应的样本信息填写,工具支持人、小鼠、大鼠三个物种样本的ATAC-Seq分析。
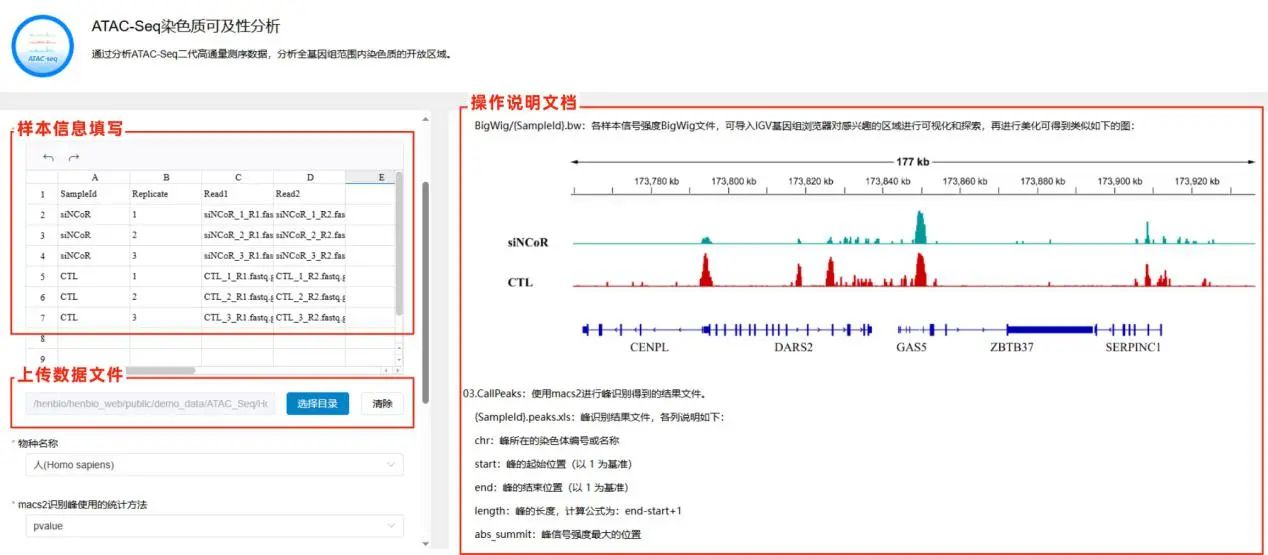
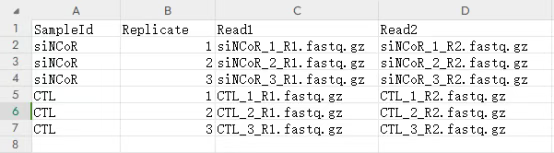
2.上传样本信息描述文件说明
(1)第一列:样本名称(SampleId)。
(2)样本重复编号:第一个重复填1,第二个重复填2,依次类推。
(2)第三列:双末端测序的第一个文件名称(Read1)。
(3)第四列:双末端测序的第二个文件名称(Read2)。
请确保Read1和Read2的文件名称正确无误,不要颠倒。您可以先在Excel中整理这些信息,然后使用复制(Ctrl+C)和粘贴(Ctrl+V)操作将数据输入到网页的表格输入框中。
如下表所示:
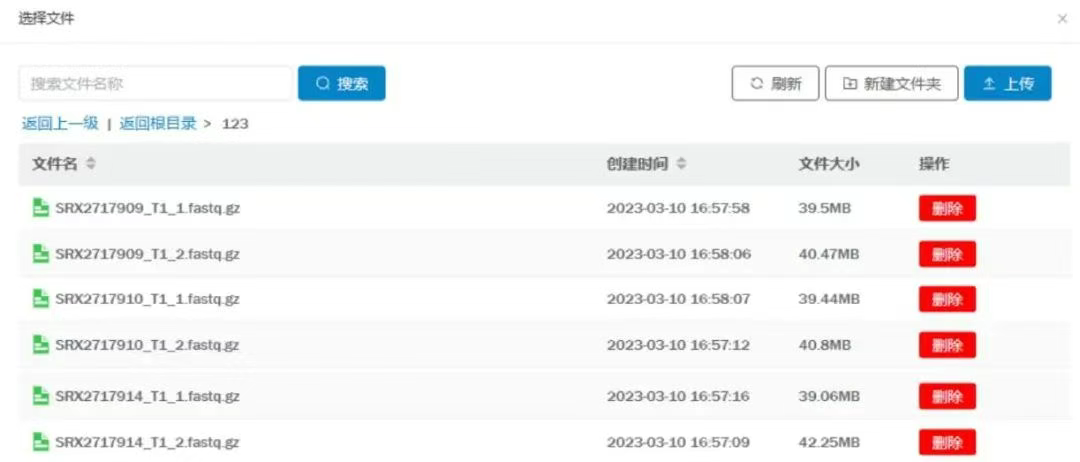
3.上传文件目录选择
数据文件所在目录:选择样本描述文件中的Read1、Read2对应的测序文件所在的目录。在分析前,您应该先把需要分析的数据文件上传到平台特定目录,确保所有需要一起分析的文件位于同一个目录里面。
4.查看消耗费用并运行
选择好参数后点击“查看总费用按钮”,页面弹出本次分析总共消耗明细,确认无误后点击“开始分析”。
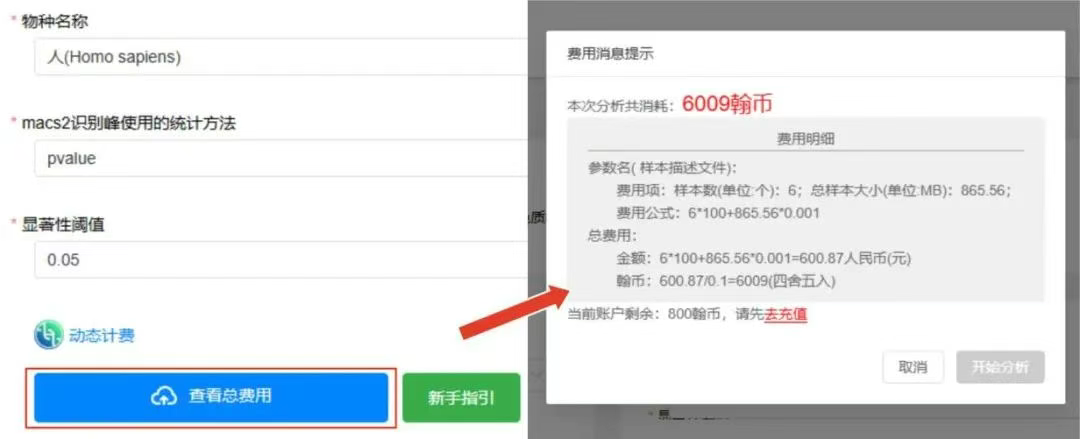
5.任务查看
点击运行后,可在我的项目中查看任务的运行情况和结果。默认情况下新任务将会在最上方展示。也可以通过项目名、任务编码、运行状态进行搜索,找到需要的任务。如下图:当状态已完成时,表示任务成功结束。
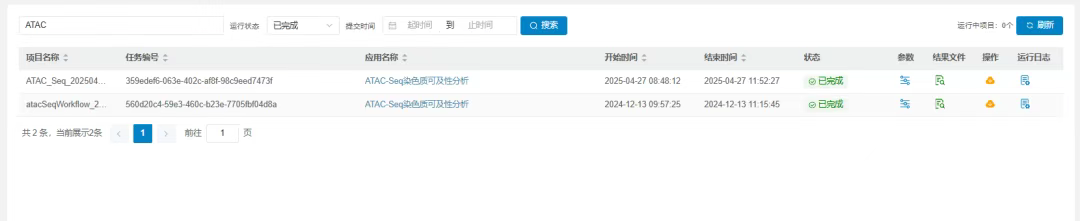
点击下载按钮可直接打包下载全部结果。
6.输出结果解读

所有结果位于ATAC_Seq_result.tar.gz的压缩文件中,解压后各目录和文件说明如下:
(1)QualityControl:质量控制分析结果。
fastp/qc_before_res.xls:原始数据各指标统计。
SampleId:样本名称。
Reads:测序文件总Reads数。
Bases(G):测序文件总碱基数。
Q20(%):测序质量达到Q20以上的占比。
Q30(%):测序质量达到Q30以上的占比。
R1_Len:双末端测序Read1文件平均测序长度。
R2_Len:双末端测序Read2文件平均测序长度。
GC(%):测序文件中的GC含量占比。
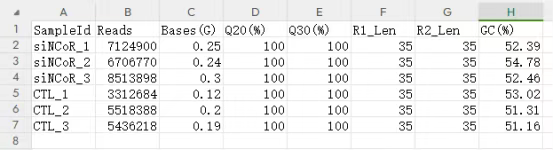
fastp/qc_after_res.xls:使用fastp软件进行去除接头、去除低质量碱基等操作后各指标统计。各字段含义和qc_before_res.xls一致。
qc_before/multiqc_report.html:原始数据各样本质控评估报告。
qc_after/multiqc_report.html:使用fastp软件进行去除接头、去除低质量碱基等操作后各样本质控评估报告。
(2)Mapping:使用Bowtie2软件把样本比对到其物种参考基因组的统计结果。
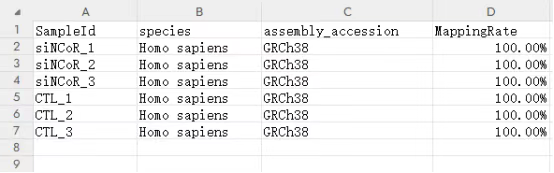
samples.mapping.rate.xls:各样本比对率统计结果,各列说明如下:
SampleId:样本名称
species:物种名称
assembly_accession:物种参考基因组版本
MappingRate:样本测序数据比对上其参考基因组的reads数占比。
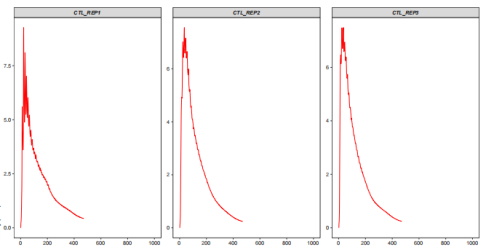
Fragment_length_distribution.pdf: 使用ATACseqQC 评估ATAC测序数据得到的样本片段大小分布图。实验比较理想的情况下会在100bp之内有一个无核小体区域的峰,200bp附近为一个核小体区域的峰,400bp附近为2个核小体区域的峰等。
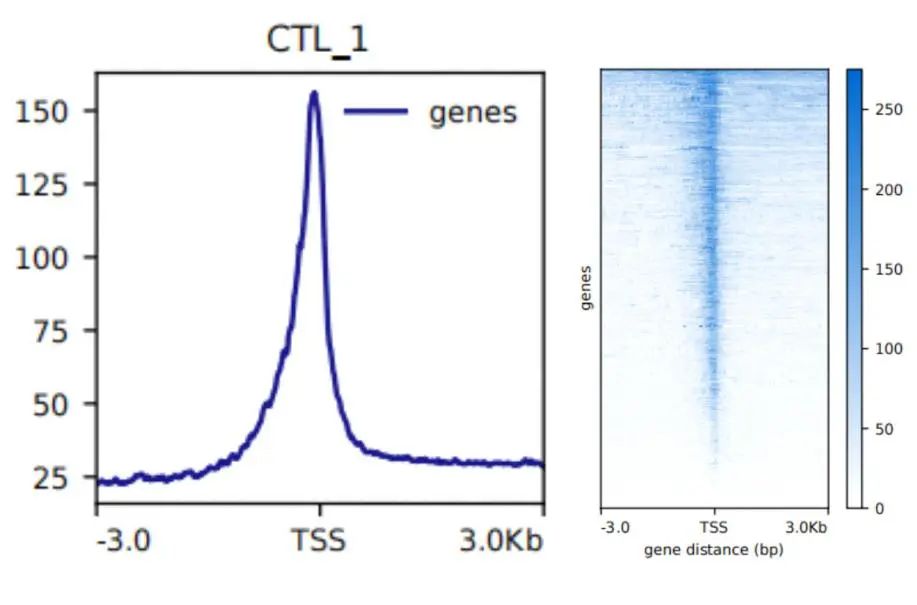
heatmap_plot/{SampleId}.TSS.heatmap.pdf:各样本在TSS上下游3kb区域内的覆盖度(信号强度)分布热图。
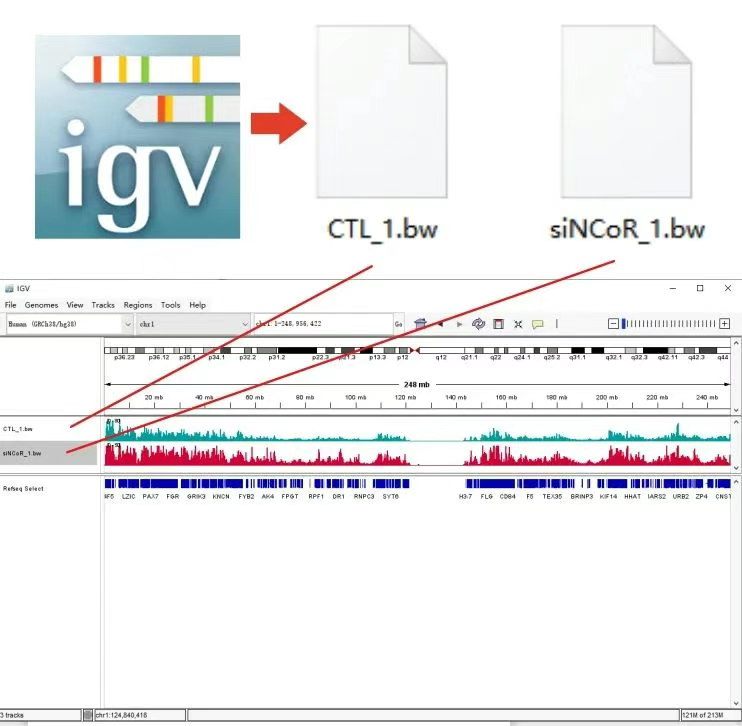
BigWig/{SampleId}.bw:各样本信号强度BigWig文件,可导入IGV基因组浏览器对感兴趣的区域进行可视化和探索,再进行美化可得到类似如下的图:
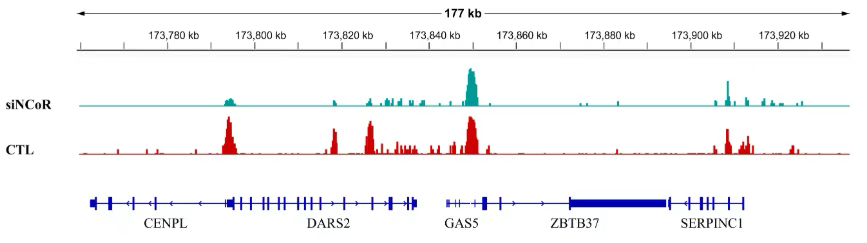
导出美化结果:
(3)CallPeaks:使用macs2进行峰识别得到的结果文件。
{SampleId}.peaks.xls:峰识别结果文件,各列说明如下:
chr:峰所在的染色体编号或名称
start:峰的起始位置(以 1 为基准)
end:峰的结束位置(以 1 为基准)
length:峰的长度,计算公式为:end-start+1
abs_summit:峰信号强度最大的位置
pileup:峰区域的信号强度(覆盖度或读段堆叠的深度)
-LOG10(pvalue):峰区域显著性检验的 P 值的对数转换值
fold_enrichment:峰信号相对于背景信号的富集倍数
-LOG10(qvalue):峰的多重检验校正后的 P 值(FDR)的对数转换值
name:峰的名称,每个峰的唯一标识符。
{SampleId}.peaks.bed:峰识别结果bed格式文件,可导入IGV可视化,坐标从0开始。
{SampleId}.summits.bed:顶峰结果文件,坐标从0开始。
(4)PeaksAnnotation:使用homer进行峰注释结果文件
{SampleId}.peaks.annotation.xls:峰注释结果文件
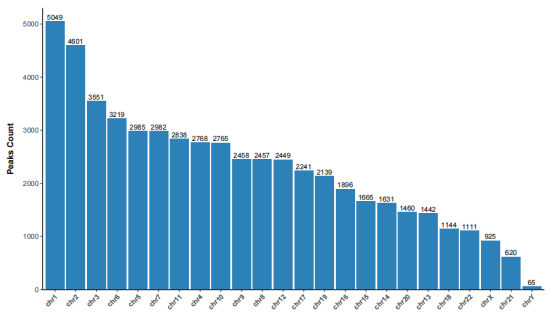
{SampleId}.peaks.annotation.barplot.pdf:富集峰在各染色体分布柱形图
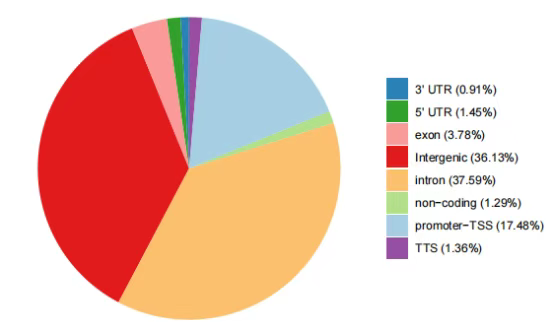
{SampleId}.peaks.annotation.pieplot.pdf:富集峰在各基因元件分布饼图
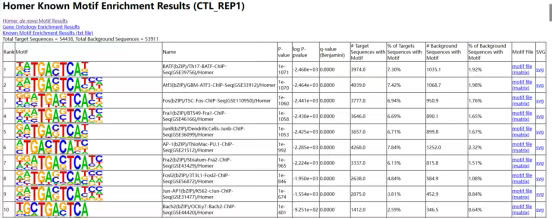
(5)findMotifs:使用homer对富集峰进行motif分析的结果文件
knownResults.html:homer分析得到的已知motif及其转录因子结果,一般P-value小于1e-50可信度才比较高
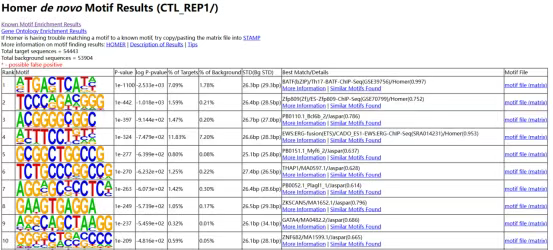
homerResults.html:homer新发现的motif结果,一般P-value小于1e-50可信度才比较高
平台入口:https://henbio.com/tools
工具链接:https://www.henbio.com/tooldetail?id=314
如果您觉得这个网站对您有帮助,还请您帮忙多多转发,与他人分享,让更多人受益。如果内容有任何侵权或是错误,恳请您及时联系我,我一定第一时间改正,感谢!

















 688
688

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









