



一、理论部分
二、实操
表型处理
使用Phenotype包(R package)来进行BLUP计算得到校正后的表型值
https://cloud.tencent.com/developer/article/1677209
EMMAX软件
EMMAX的表型数据格式
第一列,FAMID通常指的是“Family ID”的缩写
第二列,INDID代表的是个体的标识符,它是“Individual ID”的缩写,也就是他允许有重复
第三列,表型值
EMMAX的基因组数据格式
vcf文件
前几行是以"#"开头的注释信息,每行是一个SNP,每列是一个个体。行与列整合了一个群体的SNP全部变异信息。
利用plink把vcf文件转换为tped文件。
TPED文件和PED文件的主要区别在于数据的组织和展示方式。以下是具体的比较:
数据组织方式:TPED文件是把PED文件中的信息重新整合,具体做法是在MAP文件的四列之后,加上了PED文件中后面的基因型信息旋转90°之后的形式。因此,TPED文件的一行代表一个SNP的信息,而PED文件的一行代表一个样本的信息。
数据展示方式:在TPED文件中,每行代表一个SNP位点,列数取决于样本量大小,前四列内容同MAP文件,后面所有列为所有样本在该SNPs位点处的基因型信息。而在PED文件中,每行代表一个样本,前六列的内容与TFAM文件相同,后面的列为该样本在各个SNP位点处的基因型信息。
ped文件包含以下几列:
第一列:Family ID。
第二列:Individual ID。自然群体这列和Family ID是一样的。
第三列:Paternal ID。未提供信息的话这列为0。
第四列:Maternal ID。未提供信息的话这列为0。
第五列:Sex。未提供信息的话这列为0。
第六列:Phenotype。一般来说,直接拿vcf转换的话这列为-9,也就是缺失。
第七列开始就是个体在每个标记位点的基因型。
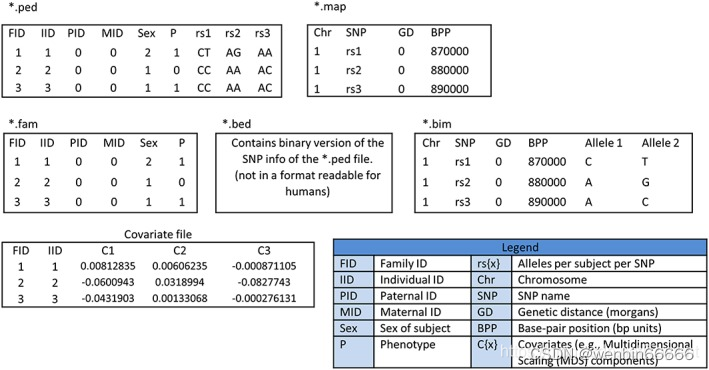
map文件包含以下几列:
第一列:染色体编号。
第二列:SNP编号。
第三列:遗传距离。未提供信息的话这列为0。
第四列:物理位置。
数据格式与其相互转换
vcf格式转换为ped格式
./plink --vcf /mnt/g/bioinfor/process/GWAS/基因型相关文件/517_plink.vcf --recode 12 --allow-extra-chr --chr-set 26 --out /mnt/g/bioinfor/process/GWAS/基因型相关文件/517
./plink是由于plink软件就在我所在的目录内,若在其他目录,需要指定路径。
–recode参数,设定输出的为为文本格式ATCG,而非二进制。–recode后还可以跟参数,设置SNP单倍型的表现方式,如双数字0/1、0/0或单数字0,1,2,具体见此链接的测试:https://www.cnblogs.com/liujiaxin2018/p/13795230.html
命令运行结束后会输出.map与.ped文件
我用了–recode 12 参数
–chr-set 26设置染色体数目
–allow-extra-chr 允许scaffold染色体
https://blog.youkuaiyun.com/Taylent/article/details/102523295
从ped格式文件中筛选出所需的材料
./plink --file /mnt/g/bioinfor/process/GWAS/基因型相关文件/517N --keep /mnt/g/bioinfor/process/GWAS/基因型相关文件/select.txt --recode 12 --out /mnt/g/bioinfor/process/GWAS/基因型相关文件/377
–keep功能 需要准备原始的被提取文件,然后是提取需要的参考文件,储存了需要提取哪些样本的信息
这里面用作筛选的select文件的保存格式需要注意,从表格导出txt文本的时候有以制表符分割和无格式文本两种,制表符分割的文件要大一些,是可以工作的,无格式文本会报错:无法读取select文件。
最后是使用PRN格式的文件成功的。
另外第一列是材料的编号,第二列也是材料的编号,只有一行是不行的。这两列在人类或哺乳动物的研究中是用来分家系的,两列可以不同,在植物学的自然群体中,设为相同。
参考:https://dengfei2013.gitee.io/plink-cookbook/plink%E6%96%87%E4%BB%B6%E6%8F%90%E5%8F%96.html#%E6%A0%B7%E6%9C%AC%E6%8F%90%E5%8F%96
ped格式转化为tped格式
./plink --file /mnt/g/bioinfor/process/GWAS/基因型相关文件/375 --recode 12 transpose --out t377 --allow-extra-chr --chr-set 26
–file 参数 只需要用户指定一个ped map 文件共有的文件前缀,它会自动读取两个文件。如果按tab补全文件名,则会出错。
–file参数可以替换为 --ped 只转换一个ped文件,这个时候补全ped文件的全名。
不需要指定输出文件的路径,默认为ped文件所在的位置
构建亲缘矩阵
./emmax-kin-intel64 /mnt/g/bioinfor/process/GWAS/基因型相关文件/t375 -v -d 10 -o t377-kin
这里输出文件的位置需要注意,我重复计算了很多遍,因为我找不到输出文件,软件说的-o参数可以指定输出路径与文件名,我发现不管怎么设置,输出文件最后都存放在./emmax-kin-intel64所处的目录,还好计算成功了。
再进一步调整数据
把不同文件的每一行都对应起来。
关联分析计算
/emmax-intel64 -v -d 10 \ #
-t snp_filter \ # 输出snp数据的前缀
-p sample.table \ # 表型数据
-k pop.kinship \ # 亲缘关系矩阵
-o emmax.out \ # 输出文件前缀
./emmax-intel64 -v -d 10 -t /mnt/g/bioinfor/process/GWAS/基因型相关文件/t375 -p /mnt/g/bioinfor/process/GWAS/表型文件/375_adjmean.txt -k /mnt/g/bioinfor/process/GWAS/基因型相关文件/t375-kin -o EMMAX_out
EMMAX 输出计算结果
整理输出结果准备绘图
EMMAX的输出文件后缀为.ps,文件是没有列名的,但是已知第一列为ID,第二列为回归系数(beta),第三列为回归系数的标准差,第四列为为P值。
由于采用的是CMplot包绘图,这个绘图需要构建一个新文件,它的格式是第一列为ID,第二列为chr,第三列position,第四列为p值。第一列的ID与染色体编号都可以从最初从517份重测序文件筛选出的375份材料的map文件,map文件第一列是染色体编号,第二列是SNP的ID,第四列是SNP的物理位置。
map_375<-read.table("G:/bioinfor/process/GWAS/基因型相关文件/375.map")#读入R
plot<-cbind(map_375[,2],map_375[,1],map_375[,4],EMMAX[,4])#重新组合出新数据
plotGWAS<-as.data.frame(plot)#把matrix数据格式转换为data frame
曼哈顿图的阈值计算
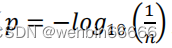
CMplot包自动计算曼哈顿图上显示的阈值,只需提供1/n即可。
CMplot(plotGWAS, threshold = threshold, amplify = F, file = "tiff", plot.type=c("m","q"))
分别绘制曼哈顿图和QQ图,存放到R的工作空间。



















 到【灌水乐园】发言
到【灌水乐园】发言








