






生信碱移
GBCD算法工具
无监督广义二元协方差分解,可以算的上是真正的单细胞患者异质性研究神器了。
单细胞RNA测序(scRNA-seq)技术在解析肿瘤转录异质性方面的分辨率远高于传统的群体转录组分析,为深入理解癌症生物学提供了新视角。通过分析转录变异模式,可识别基因表达程序(GEP),即一组具有协同转录变化的基因。这些GEPs可能具有患者特异性,也可能在部分患者间共享。共享的GEPs通常与分子亚型或细胞状态相关,从而揭示疾病病因。
在单细胞领域,降维分解是绕不过的分析流程。其主要是指将高维基因表达汇总成一个个具有代表性的特征组分,比如最常见的主成分分析(PCA) 便是将基因表达矩阵分解成了一个个主成分以及相应的载荷矩阵 (表示基因对不同主成分的贡献)。小编先前分享了多篇基于降维分解的方法,比较有代表性的是①近期发表在 Nature Biotechnology 的单细胞可解释张量分解算法 scITD,以及②登上 Nature Protocols 的热门算法贝叶斯非负矩阵分解 CoGAPS,感兴趣的铁子可以点击下方相应链接学习。
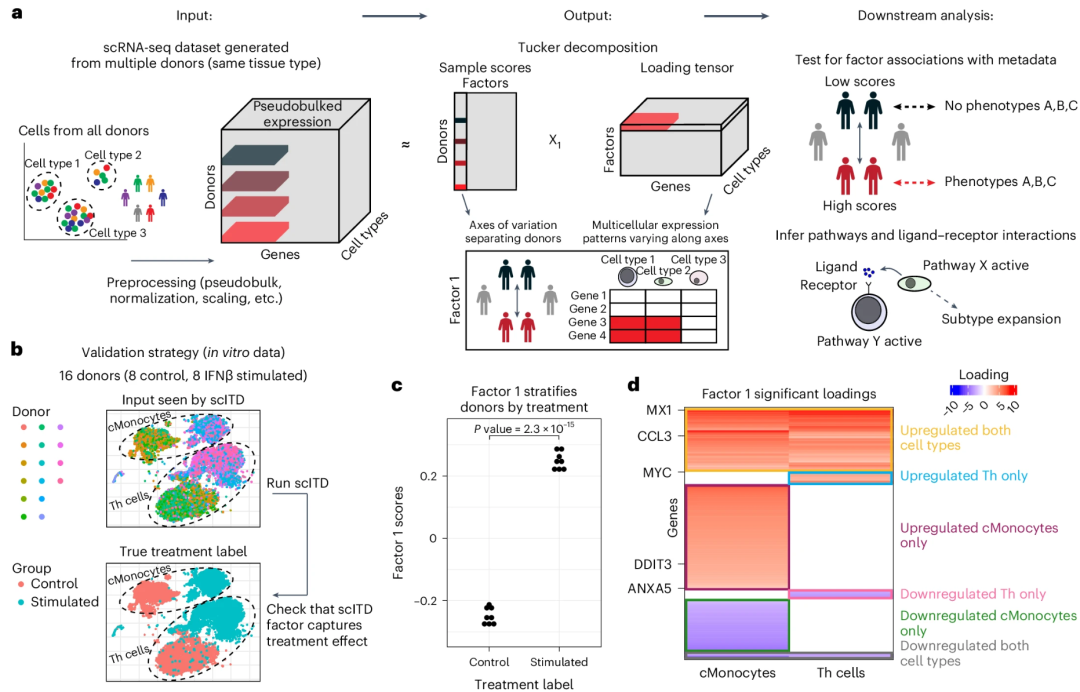
▲ 图:scITD 的设计概述及功能演示。简单来讲,scITD针对每种细胞类型生成“供体×基因”的伪批量表达矩阵(这里的供体就是个体)。①这些矩阵通过细胞类型维度堆叠,形成一个三维张量(供体×基因×细胞类型)。②该张量在进一步分析之前会经过标准化、归一化和缩放等预处理步骤。③随后,作者使用Tucker张量分解将三维数据分解为几个因子矩阵(样本因子、基因因子、细胞类型因子)和一个核心张量。Tucker张量分解类似于PCA,但它是针对三维数据的。点击此处跳转
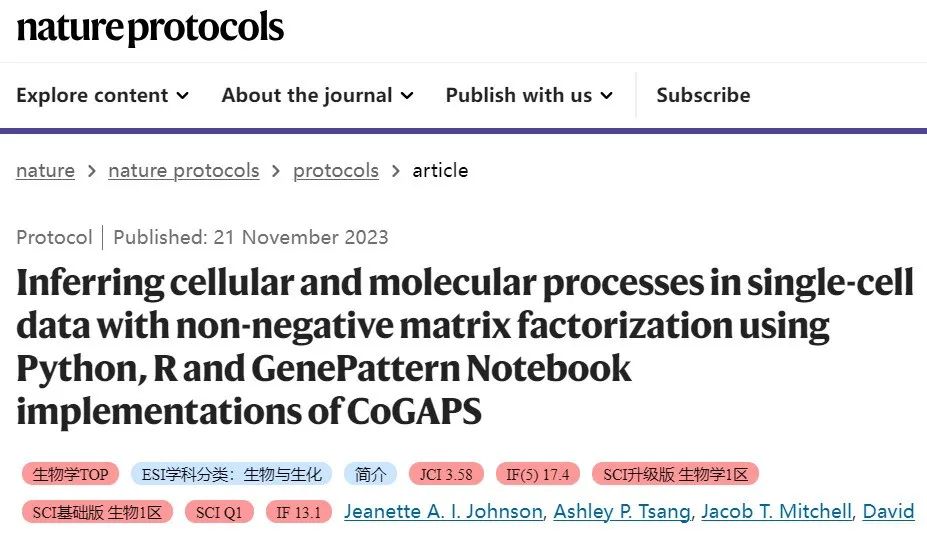
▲ 图:来自美国约翰霍普金斯大学的研究者分享的一套流程。介绍了贝叶斯NMF算法(CoGAPS),并演示了如何通过 CoGAPS 分析量化公共领域单细胞 RNA 测序数据中的细胞状态转换。点击此处跳转
尽管前面已经开发了一些方法,但是在肿瘤学等疾病研究领域,由于患者异质性的存在,使得识别患者共享的基因表达程序GEPs仍然是一个非常大的挑战。比如,在肿瘤当中,患者的异质性被称为肿瘤异质性。在包含多个肿瘤的 scRNA-seq 数据中,恶性细胞通常按患者聚类,而患者间的重叠很少。这种强烈的患者间异质性往往掩盖了肿瘤间共享的、更微妙但重要的异质性模式。不仅如此,针对多个数据集的分析还面临着批次效应的问题,批次效应往往还与患者异质性混淆,大多算法甚至在批次矫正时会去除这些有意义的生物学信号。
为此,来自芝加哥大学的研究者提出了一种基于矩阵分解的新方法 GBCD (广义二值协方差分解),于2025年1月2号见刊于 Nature Genetics [IF:31.7]。作者的实验结果表明,GBCD 克服了现有方法的局限性,能够在强肿瘤间异质性下取得更具解释性的结果。

▲ 在文章的演示中,GBCD能够在 HNSCC 数据中识别出与亚型相关的 GEPs,在 PDAC 数据中精细化临床亚型,并发现与应激响应通路相关的 GEP并独立预测较差生存率。DOI: 10.1038/s41588-024-01997-z。
本文将简要介绍GBCD的方法原理以及代码示例运行,感兴趣的铁子可以查阅以下链接:
-
https://github.com/stephenslab/gbcd
GBCD 原理
GBCD是一个无监督方法,这意味着其并不会过于利用现有的样本标签,混淆样本批次或肿瘤异质性信息。简单来讲,GBCD 通过将单细胞基因表达矩阵分解为细胞与模式的关联矩阵(模式特指前面所提到的基因表达程序GEP) 和基因与模式的贡献矩阵。其数学描述为:
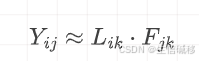
上面公式非常简练:

"![]() " 指代该分解是一个优化问题,其主要基于近似二值化和正交性的两个假设来优化矩阵分解结果:
" 指代该分解是一个优化问题,其主要基于近似二值化和正交性的两个假设来优化矩阵分解结果:
-
① 二值化假设:
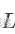 数值范围限定接近于0或1,其目标是捕捉任何的离散结构,例如患者效应或肿瘤亚型。这种假设被作者称为广义二值假设(Generalized Binary Assumption)。
数值范围限定接近于0或1,其目标是捕捉任何的离散结构,例如患者效应或肿瘤亚型。这种假设被作者称为广义二值假设(Generalized Binary Assumption)。 -
② 正交性假设:限制
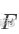 的列彼此正交 (正交其实是一个常见的数学名词,不同的铁子可以自行谷歌),简单来说就是确保不同的模式不会混在一起。
的列彼此正交 (正交其实是一个常见的数学名词,不同的铁子可以自行谷歌),简单来说就是确保不同的模式不会混在一起。
小编给没听懂的同学总结一下,通过 GBCD 我们最终可以获得细胞与模式关联矩阵![]() 、基因与模式的关联矩阵
、基因与模式的关联矩阵![]() ,前者可以用于确定细胞属于哪些特定模式 (区分不同细胞群体以及细胞亚型),后者可以用于确定哪些基因在特定生物学模式中上调或下调
,前者可以用于确定细胞属于哪些特定模式 (区分不同细胞群体以及细胞亚型),后者可以用于确定哪些基因在特定生物学模式中上调或下调
GBCD 示例运行
代码运行讲解可以分为以下六个步骤:
① GBCD基于R编程语言实现,可以通过以下代码安装:
# install.packages("remotes")
remotes::install_github("stephenslab/gbcd")
运行示例还需要一些其它的R包,可以一起引用 (没有引用的话需要安装):
library(Matrix)
library(ggplot2)
library(cowplot)
library(ggrepel)
library(pheatmap)
library(Rtsne)
library(flashier)
library(gbcd)
#
library(GBCD)
② 内置数据集介绍:来源于头颈癌(HNSCC)患者的基因表达数据,包含了 2176 个恶性细胞,hnscc$info为细胞注释信息,其中:
-
subtype列:代表预先知道的每个细胞的亚型 -
sample列:代表相应的样本 -
cell.type列:代表相应的细胞注释
data(hnscc)
dim(hnscc$Y)
head(hnscc$info)
# [1] 2176 17113
# cell.id lymph.node cancer.cell
# HN26_P14_D11_S239_comb HN26_P14_D11_S239_comb 1 1
# HN26_P25_H09_S189_comb HN26_P25_H09_S189_comb 1 1
# HN26_P14_H06_S282_comb HN26_P14_H06_S282_comb 1 1
# HN25_P25_C04_S316_comb HN25_P25_C04_S316_comb 1 1
# HN26_P25_C09_S129_comb HN26_P25_C09_S129_comb 1 1
# HNSCC26_P24_H05_S377_comb HNSCC26_P24_H05_S377_comb 1 1
# cell.type patient.id sample subtype
# HN26_P14_D11_S239_comb 0 T26 MEEI26_LN Atypical
# HN26_P25_H09_S189_comb 0 T26 MEEI26_LN Atypical
# HN26_P14_H06_S282_comb 0 T26 MEEI26_LN Atypical
# HN25_P25_C04_S316_comb 0 T25 MEEI25_LN Basal
# HN26_P25_C09_S129_comb 0 T26 MEEI26_LN Atypical
# HNSCC26_P24_H05_S377_comb 0 T26 MEEI26_LN Atypical
③ 可视化HNSCC数据集,可以发现存在明显的肿瘤异质性或者是批次效应:
set.seed(1)
cols <- order(apply(hnscc$Y,2,sd),decreasing = TRUE)
cols <- cols[1:3000]
res <- Rtsne(as.matrix(hnscc$Y[,cols]),normalize = TRUE)
colnames(res$Y) <- c("tsne1","tsne2")
pdat <- cbind(res$Y,hnscc$info)
ggplot(pdat,aes(x = tsne1,y = tsne2,color = sample,shape = subtype)) +
geom_point(size = 1) + scale_color_manual(values = hnscc$sample_col) +
scale_shape_manual(values = c(15,16,18)) +
theme_cowplot(font_size = 10)
如上图所示,细胞间存在强烈的肿瘤间异质性。图中显示出典型样本分群,即细胞的 t-SNE 可视化的主要结构是按患者聚类。
④ 使用fit_gbcd函数进行GBCD分析:
res.gbcd <- fit_gbcd(Y = hnscc$Y, Kmax = 12, maxiter1 = 100,
maxiter2 = 50, maxiter3 = 50,
prior = flash_ebnm(prior_family = "generalized_binary",
scale = 0.04))
# [1] "Form cell by cell covariance matrix..."
# user system elapsed
# 15.207 0.072 16.125
# [1] "Initialize GEP membership matrix L..."
# Adding factor 1 to flash object...
# Wrapping up...
# Done.
# Adding factor 2 to flash object...
# Adding factor 3 to flash object...
# Adding factor 4 to flash object...
# Adding factor 5 to flash object...
# Adding factor 6 to flash object...
# Adding factor 7 to flash object...
# Adding factor 8 to flash object...
# Adding factor 9 to flash object...
# Adding factor 10 to flash object...
# Adding factor 11 to flash object...
# Adding factor 12 to flash object...
# Wrapping up...
# Done.
# Backfitting 12 factors (tolerance: 7.06e-02)...
# Difference between iterations is within 1.0e+04...
# Difference between iterations is within 1.0e+03...
# Difference between iterations is within 1.0e+02...
# --Maximum number of iterations reached!
# Wrapping up...
# Done.
# Backfitting 12 factors (tolerance: 7.06e-02)...
# Difference between iterations is within 1.0e+04...
# Difference between iterations is within 1.0e+03...
# Difference between iterations is within 1.0e+02...
# Difference between iterations is within 1.0e+01...
# --Maximum number of iterations reached!
# Wrapping up...
# Done.
# user system elapsed
# 35.554 2.770 39.815
# [1] "Estimate GEP membership matrix L..."
# Backfitting 23 factors (tolerance: 7.06e-02)...
# Difference between iterations is within 1.0e+05...
# Difference between iterations is within 1.0e+04...
# Difference between iterations is within 1.0e+03...
# --Maximum number of iterations reached!
# Wrapping up...
# Done.
# Backfitting 23 factors (tolerance: 7.06e-02)...
# Difference between iterations is within 1.0e+03...
# Difference between iterations is within 1.0e+02...
# --Maximum number of iterations reached!
# Wrapping up...
# Done.
# user system elapsed
# 64.059 3.096 67.728
# [1] "Estimate GEP signature matrix F..."
# Backfitting 23 factors (tolerance: 5.55e-01)...
# Difference between iterations is within 1.0e+03...
# Difference between iterations is within 1.0e+02...
# --Maximum number of iterations reached!
# Wrapping up...
# Done.
# user system elapsed
# 314.585 13.651 331.738
需要注意的是该函数的参数设置:
-
Kmax:模式数量的上限,上一个原理处所提到的k。需要注意,设置的 Kmax 只是初始化的目的,最终 GEP 数量通常接近但不一定等于 (并且可能大于) Kmax。根据作者的经验,Kmax 在 10 到 40 之间通常效果良好。 -
scale:每个条目分配的广义二进制先验,默认是0.04。一般来说,对于包含更多细胞的大型数据集,较小的比率是推荐的,0.01 到 0.1 之间的值通常效果很好。 -
prior:用于为GEP指定其他非负先验,例如,用户可以通过在调用fit_gbcd时设置prior = ebnm_point_exponential来指定点指数先验。但是,作者的经验表明,GB先验通常在解析多个样本和/或研究中的肿瘤转录异质性方面表现最佳。 -
此外,可以看到这个方法不需要提供细胞的患者/数据集身份信息,纯纯的非监督方法。
⑤ 细胞与模式关联得分矩阵![]() 的可视化(只是小编的称呼,英文为GEP membership estimates)。通过
的可视化(只是小编的称呼,英文为GEP membership estimates)。通过$L提取,可以看到其是不同的基因表达模式GEP在细胞中的得分 (如前所述 0-1 范围)。
head(round(res.gbcd$L, 3))
# Baseline GEP1 GEP2 GEP3 GEP4 GEP5 GEP6 GEP7
# HN26_P14_D11_S239_comb 0.735 0 0.000 0.895 0.354 0.000 0.000 0.573
# HN26_P25_H09_S189_comb 0.657 0 0.141 0.776 0.000 0.479 0.000 0.617
# HN26_P14_H06_S282_comb 0.791 0 0.244 0.710 0.606 0.000 0.000 0.713
# HN25_P25_C04_S316_comb 0.412 0 0.661 0.212 0.000 0.649 0.000 0.000
# HN26_P25_C09_S129_comb 0.645 0 0.000 0.743 0.000 0.382 0.000 0.733
# HNSCC26_P24_H05_S377_comb 0.586 0 0.000 0.739 0.000 0.405 0.345 0.696
# GEP8 GEP9 GEP10 GEP11 GEP12 GEP13 GEP14 GEP15 GEP16
# HN26_P14_D11_S239_comb 0 0.000 0.664 0 0 0.437 0.000 0.000 0.000
# HN26_P25_H09_S189_comb 0 0.000 0.000 0 0 0.471 0.000 0.591 0.000
# HN26_P14_H06_S282_comb 0 0.000 0.000 0 0 0.452 0.000 0.000 0.585
# HN25_P25_C04_S316_comb 0 0.695 0.672 0 0 0.606 0.793 0.670 0.000
# HN26_P25_C09_S129_comb 0 0.000 0.000 0 0 0.000 0.000 0.748 0.000
# HNSCC26_P24_H05_S377_comb 0 0.000 0.000 0 0 0.000 0.461 0.586 0.000
# GEP17 GEP18 GEP19 GEP20
# HN26_P14_D11_S239_comb 0 0.00 0 0
# HN26_P25_H09_S189_comb 0 0.00 0 0
# HN26_P14_H06_S282_comb 0 0.36 0 0
# HN25_P25_C04_S316_comb 0 0.00 0 0
# HN26_P25_C09_S129_comb 0 0.00 0 0
# HNSCC26_P24_H05_S377_comb 0 0.00 0 0
用热图可视化看看呢:
# 创建注释信息
anno <- data.frame(sample = hnscc$info$sample, subtype = hnscc$info$subtype)
rownames(anno) <- rownames(res.gbcd$L)
anno_colors <- list(sample = hnscc$sample_col, subtype = hnscc$subtype_col)
cols <- colorRampPalette(c("gray96", "red"))(50)
brks <- seq(0, 1, 0.02)
# 可视化
pheatmap(res.gbcd$L[order(anno$sample), -c(1)], cluster_rows = FALSE,
cluster_cols = FALSE, show_rownames = FALSE, annotation_row = anno,
annotation_colors = anno_colors, annotation_names_row = FALSE,
angle_col = 45, fontsize = 9, color = cols, breaks = brks,
main = "")
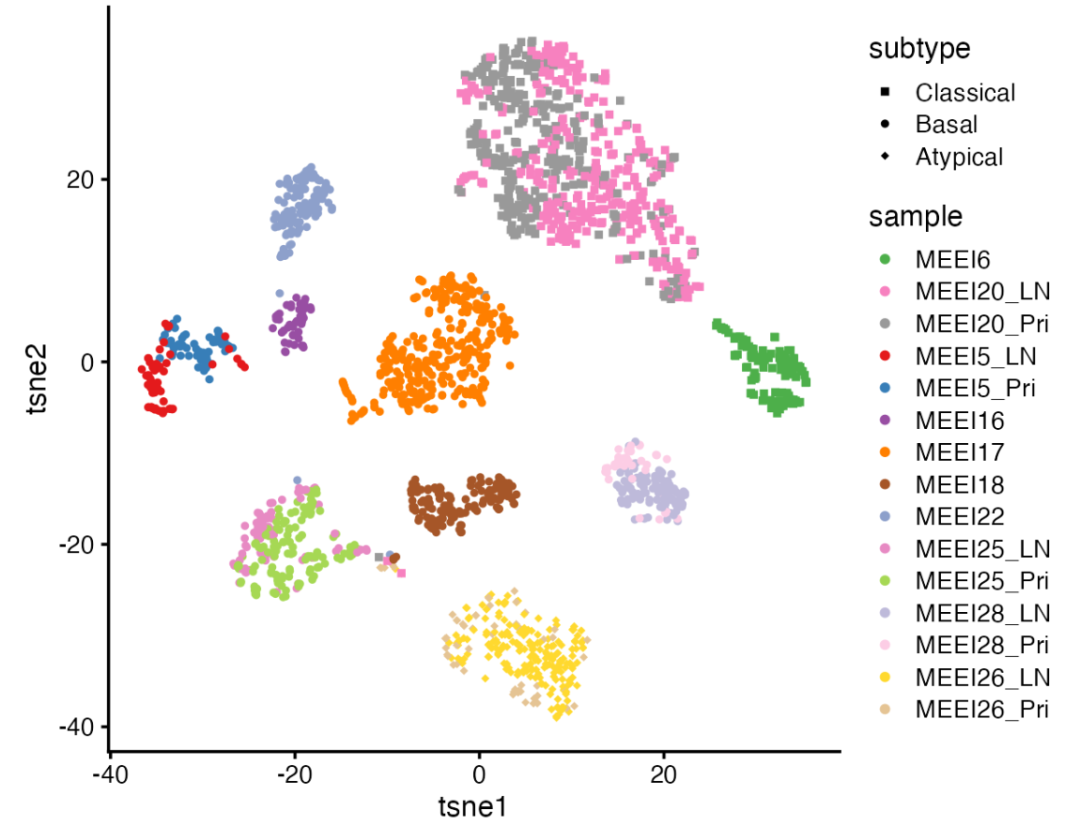
▲ 热图显示了 2,176 个细胞(行)的模式得分关系。细胞按照肿瘤分子亚型从上到下排列,并按原患者来源排列。得分针对每个 GEP 分别重新缩放,使得每个 GEP 的最大得分始终为 1。
从上述热图中,①首先我们可以看到GEPs 1-3与之前定义的分子亚型高度一致:GEP1主要在2例经典患者 (Classical) 的细胞中活跃;GEP2主要在7例基底细胞患者 (Basal) 的细胞中活跃;而GEP3主要在1例非典型 (Atypical) 患者的细胞中活跃。这表明 GBCD 可以从单细胞数据中独立提取分子亚型信息;②在其他 GEPs 中,有些主要在个别患者中活跃,因此可以解释为患者特异性 GEPs,如 GEPs 8、9 和 11;其他 GEPs 则在不同患者之间共享,但不对应于分子亚型。每个 GEP 的患者特异性与共享表达程度可以通过gep_annotation函数量化。效果还是很牛的,要知道这可是无监督的方法!
⑥ 基因与模式关联得分矩阵![]() 的可视化(只是小编的称呼,英文为GEP signature estimates )。通过
的可视化(只是小编的称呼,英文为GEP signature estimates )。通过$F$lfc提取,可以看到其是不同的基因对于基因表达模式GEP的得分。每个条目大约表示为属于特定 GEP 相关的对数折叠变化(以 2 为底)。
head(round(res.gbcd$F$lfc, 3))
# Baseline GEP1 GEP2 GEP3 GEP4 GEP5 GEP6 GEP7 GEP8
# C9orf152 0.000 0.064 0.000 0.004 0.000 0.003 0.000 0.000 0.000
# ELMO2 2.056 0.000 -0.466 0.715 0.003 -0.273 -0.065 0.015 -0.051
# PNMA1 1.933 0.003 -1.326 0.035 -0.002 0.019 -0.043 -0.019 -0.018
# MMP2 0.763 -0.468 2.441 -0.897 -0.005 0.786 -0.597 -0.004 -1.206
# TMEM216 2.141 1.046 -0.652 0.021 0.006 -0.012 -0.115 -0.021 -0.002
# TRAF3IP2-AS1 0.216 0.293 0.000 0.588 0.001 0.132 0.000 0.003 0.000
# GEP9 GEP10 GEP11 GEP12 GEP13 GEP14 GEP15 GEP16 GEP17
# C9orf152 0.000 -0.003 -0.004 0.000 0.002 0.084 0.000 0.000 0.000
# ELMO2 0.081 0.005 -0.764 -0.036 0.348 -0.352 -0.566 -0.019 -0.003
# PNMA1 2.154 0.065 -0.017 0.339 0.731 0.012 -0.048 0.014 -0.002
# MMP2 0.051 -0.031 0.026 -0.867 0.800 0.348 -0.508 0.035 -0.297
# TMEM216 1.137 -0.088 -0.401 -0.262 0.189 -0.605 -0.013 -0.429 -0.055
# TRAF3IP2-AS1 0.005 0.013 0.011 -0.004 0.025 0.449 -0.002 0.019 -0.001
# GEP18 GEP19 GEP20
# C9orf152 0.014 -0.001 0.000
# ELMO2 -0.104 0.060 0.019
# PNMA1 -0.007 -0.008 0.022
# MMP2 -1.498 -0.020 3.791
# TMEM216 -0.037 -0.012 0.029
# TRAF3IP2-AS1 -0.002 0.042 -0.001
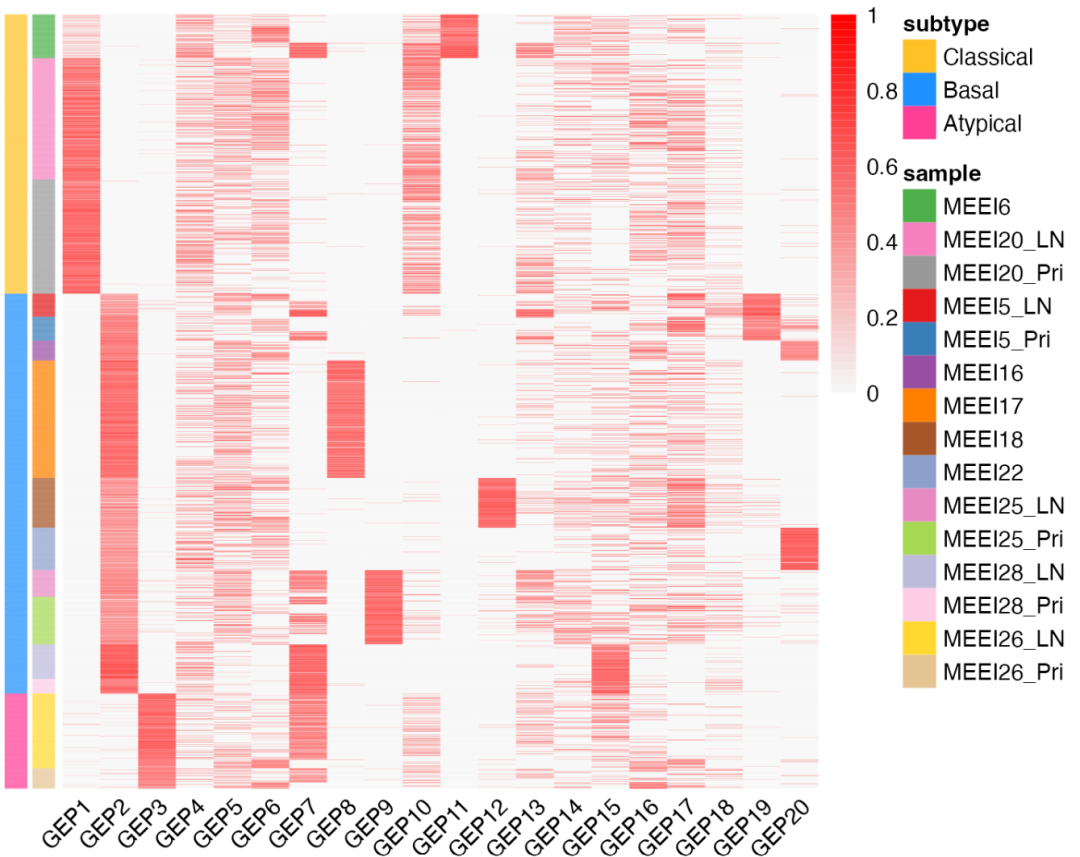
用火山图可视化呢,以GEP2这个基因表达模式为例:
# 可以看到,下面利用了 fit_gbcd 调用输出的后验统计量,包括 z 分数和局部假阳性率(lfsr)
pdat <- data.frame(gene = rownames(res.gbcd$F$lfc),
lfc = res.gbcd$F$lfc[, "GEP2"],
z = abs(res.gbcd$F$z_score[, "GEP2"]),
lfsr = res.gbcd$F$lfsr[, "GEP2"],
stringsAsFactors = FALSE)
pdat <- transform(pdat, lfsr = cut(lfsr, c(-1, 0.001, 0.01, 0.05, Inf)))
rows <- with(pdat, which(!(abs(lfc) > quantile(abs(lfc), 0.998) | (z > 10))))
pdat[rows, "gene"] <- ""
ggplot(pdat, aes(x = lfc, y = z, color = lfsr, label = gene)) + geom_point() +
geom_text_repel(color = "black", size = 2.5, segment.color = "black",
segment.size = 0.25, min.segment.length = 0,
max.overlaps = Inf, na.rm = TRUE) +
scale_color_manual(values = c("coral", "orange", "gold", "deepskyblue")) +
labs(x = "log-fold change", y = "|posterior z-score|") +
guides(colour = guide_legend(override.aes = list(size = 2))) +
theme(plot.title = element_text(hjust = 0.5,size = 12),
axis.text = element_text(size = 10),
axis.title = element_text(size = 10),
legend.title = element_text(size = 12),
legend.text = element_text(size = 10),
legend.position = "bottom") +
ggtitle("Volcano plot of gene signature for GEP2")
▲ 上图可以理解为每个基因的贡献,使用这个结果也可以对不同的基因表达模式得分计算相应的GSEA通路富集指标奥。
真是江山更有才人出啊
好好学习学习
今天就分享到这里了


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









