








2022就已经预打印了,这次算老文新看。
题目:Score-based generative modeling for de novo protein design
文献来源:https://doi.org/10.1038/s43588-023-00440-3(Nature Computational Science)
代码:https://gitlab.com/mjslee0921/proteinsgm
内容:
具有预先防御功能和特性的从头蛋白质结构的生成仍然是蛋白质设计中的一个具有挑战性的问题。扩散模型,也被称为基于分数的生成模型(SGMs),最近在图像合成中表现出了惊人的作用。在这里,作者使用基于图像的蛋白质结构表示来开发ProteinSGM,一种基于分数的生成模型,来产生真实的从头蛋白质。通过无条件生成,作者证明了ProteinSGM可以生成类似天然的蛋白质结构,超过了以前报道的生成模型的性能。作者通过实验验证了一些从头设计,并观察到蛋白质二级结构组成与生成的主链相一致。最后,作者将条件生成应用于蛋白质的从头设计,将其作为一个图像内绘制问题,允许精确和模块化的蛋白质结构设计。
1.背景介绍
蛋白质设计存在一个很大程度上未被解决的问题,那就是设计新的骨架:我们是否可以通过蛋白质序列来生成合适的骨架。或者更深一步:我们是否可以发现那些不在天然结构中的骨架。换句话说:我们是否可以发现那些不在SCOP或者CATH数据库中的折叠骨架?由这个问题延伸而来的一个任务就是基于条件的骨架生成任务:当给定一个功能性位点的时候,我们是否可以生成保留其活性的一些兼容性的骨架?在本文中,作者想要通过深度学习来探索这个问题。
扩散模型在很多领域都取得了不少的成就。该模型定义了一个正向的扩散过程并且在数据中加入噪声,并且学习反向过程-将随机的高斯噪声从数据分布的样本中取出。而基于分数的生成模型就是扩散模型的一种类型。它在使用噪声条件神经网络在不同噪声尺度下的扰动数据估计了数据分数,或者是相对于数据的对数概率密度的梯度。本文提出的ProteinSGM,一个基于连续时间评分的生成模型(SGM),它可以生成高质量的全新蛋白。ProteinSGM生成四个完全描述蛋白质主干的矩阵,这些矩阵在罗塞塔最小化协议中被用作平滑的谐波约束。采用固定主链快速设计进行序列和转子设计,最后在约束下放松结构,生成低能量的全原子结构。作者表明,ProteinSGM产生不同长度的结构。这些结构的每个氨基酸残基的平均Rosetta energy units (REU)<–3.9 。
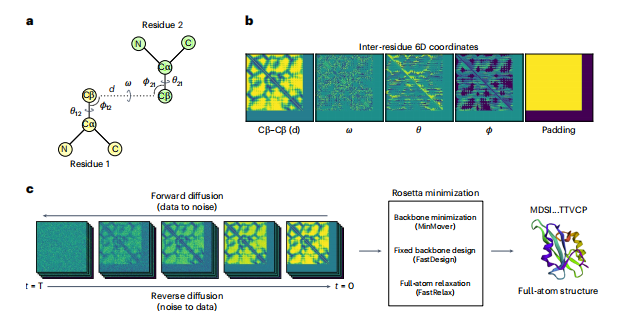
图1 模型概述图。a:两个残基间的6D坐标为d、ω、θ、ϕ。b:输入特征(6D坐标和填充通道)用于描述给定的蛋白质结构,其中6D坐标表示在-1(绿色)和1(黄色)之间归一化的距离和角值。c:给定了干扰数据对噪声的正向扩散过程,一个扩散模型从通过学习一个反向去噪过程从噪音(连续时间情况下的步长 t = T, T = 1 )中生成真实的例子(timestep t = 0)。生成的6D坐标用Rosetta作为约束最小化的输入,进行固定主干设计和全原子松弛,得到与6D坐标约束对应的蛋白质结构。
在这项工作中,作者使用蛋白质结构的类图像表示来训练一个SGM,其中每个蛋白质主干由trRosetta中定义的残基间6D坐标(图1a)表示。简而言之,模型从每个蛋白质中计算出四个对应的矩阵:Cβ-Cβ距离(以下简称d);ω和θ扭转角;以及完全描述蛋白质主干的ϕ平面角。这些矩阵构成了6D坐标,因为ϕ和θ在残基对之间是不对称的。例如,给定两个残基i和j,其中i ≠ j,ϕij≠ϕji和θij ≠ θji。为了生成不同长度的蛋白质,作者还添加了一个填充通道,表示6D坐标的边界,可以根据模型的条件来生成固定长度的蛋白质。综上所述,一个蛋白质结构表示为128×128×5张量,4个通道对应6D坐标,一个填充通道(图1b)。本文使用基于分数的生成建模的连续时间框架与随机微分方程(SDEs)(图1c)。通过估计分数函数,训练模型从高斯先验中去噪真实的6D坐标,该分数函数用于求解将高斯噪声映射到数据中的反向时间SDE。然后对生成的6D坐标进行 Rosetta最小化,使用MinMover进行带约束的主干最小化,快速设计固定主链序列和转子设计,最后使用Fastrelax约束松弛步骤生成低能量全原子结构。经过模型训练后作者评估了模型在无条件生成方面的性能,以评估样本的多样性和合理性,并通过输入掩蔽输入特征来进行条件生成。
2.数据与模型
2.1 数据
本文使用CATH 4.3 40%的序列相似性聚类来减少特定结构的冗余和潜在偏差。在对40到128之间的结构长度进行过滤后,作者得到了14,987个结构用于95:5的训练:测试分割。请注意,本文没有使用实验分辨的β-碳坐标,而是根据给定的氮、α-碳和碳坐标来推断β-碳的位置。这也有助于对没有β-碳的甘氨酸进行建模。PDB文件使用Biopython35和Biotite进行解析。
2.2 基于分数的生成建模
对于这项工作,作者使用了具有SDE的SGM的连续时间框架。
当给定一些数据X以及时间∈[0,1],由以下通用形式的SDE定义正向噪音过程:
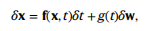
式中,f(⋅,t)为漂移系数,g (t)为扩散系数,δw为高斯白噪声。直观地说,给定一个小的时间步长δt,数据X加入具有均值f(x,t)δt和方差g2(t)δt的正态分布随机值噪声。给定一个正向SDE,可以制定一个相应的反向时间SDE,这需要了解分数∇x logpt(x),或对数概率密度相对于数据的梯度,定义如下:
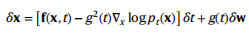
因此,一旦我们定义了前向噪声过程并学习一个近似的分数函数,我们就可以求解反向时间SDE,并将先验分布的随机样本去噪到真实样本中。作者使用与分数匹配目标对应的 variance exploding SDE discretization与Langevin Markov chain Monte Carlo(MCMC)抽样,并由以下SDE定义:
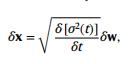
其中,σ(t) = σmin(σmax/σmin)t分别给出了下界和上界噪声方差超参数σmin和σmax。为了解决反向时间的SDE,作者估计了分数∇x logpt(x)和一个评分网络 sθ(x, t)。分数网络采用加权去噪分数匹配目标进行训练,定义如下:
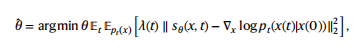
其中λ(t)是一个正的加权函数,pt(x(t)∣x(0))对应于一个扰动核,它将干净的样本x (0)扰动到有噪声的样本x (t)。
2.3 Rosetta 最小化
为了从残差间的6D坐标中获得结构,本文使用了trRosetta最小化协议。trRosetta和先前报道的基于变分自编码器的方法24使用分布图-或每个距离/角度箱的概率-来拟合一个样条函数,以生成罗塞塔最小化的平滑约束。但是,ProteinSGM直接生成距离值和角值。然后,作者使用d和ϕ的谐波函数,以及ω和θ的循环谐波函数,其平均值对应于生成的值,d的标准差为2.0埃,ϕ、ω和θ的标准差为10°。这允许在给定一组6D坐标的情况下可重复地生成结构,同时仍然放松足够的约束以生成真实的结构。
其他内容的具体阐述可见文章Method部分。
3.无条件生成
3.1 6D坐标分析
蛋白质的相邻残基限制了残基间的内部坐标,因此表现出特定的残基间分布。为了验证该模型能够学习蛋白质的自然生物物理约束并有效地捕获这些分布,作者采用完全训练的模型生成了1068个样本(长度在40aa到128aa之间,每个长度有10个样例生成)并将相邻残差的6D坐标分布与测试数据的分布进行了比较(图2)。在所有的d、ω、θ、ϕ分布中,作者观察到这些分布与测试集的分布非常匹配,这表明该模型已经学会了生成不同长度的真实的6D坐标。他们还分析了6D坐标之间的联合分布,并观察到真实样本和生成样本的相邻剩余特征在所有二维分布中具有一致性。
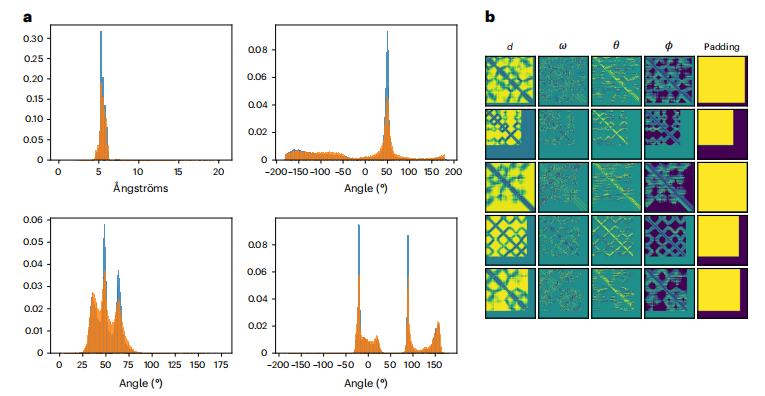
图2 6D坐标分析。a: 作者用该模型生成了1068个样本,并将其与训练集中的特征进行了比较。真实(蓝色)、ω(右上)、ω(左上)、θ(左下)和ϕ(右下)的分布有很大的重叠,表明该模型已经学习了残差6D坐标的原生约束。b,生成的矩阵示例,每一行对应单个蛋白质的特征,遵循图1所示的配色方案。
2.2 结构分析
首先,为了验证Rosetta协议在从6D坐标中可重复生成结构方面的稳定性,作者从测试集中的750个结构中提取了6D坐标,并分析了Cα RMSD。在每个最小化步骤上(补充图2)。我们观察到,在FastRelax后,平均Cα RMSD为0.97 A,这表明该方案非常适合于在<1 A精度下的可重复生成。
作者继续使用Rosetta协议生成了所有1,068个样本的全原子结构,并将它们的性质与来自测试集中的最小化结构进行了比较。他们观察到,生成的结构的Rosetta能量与原生结构几乎相同,原子的平均能量值均为-3.9REU(图3a),这明显高于最近基于变分自动编码器的方法-该报告的平均绝对REU为0.061。为了检测样本的泛化和多样性,作者在每个生成的样本与来自训练集的结构之间使用TM-align25测量TM-分数(图3b)。实验观察到,这些结构的一个子集获得了<0.5的TM-分数,证明模型生成了在训练集中不存在的新的结构。此外,这也表明ProteinSGM并不简单地记忆在训练集中发现的结构,而是极大地使最大TM-分数分布倾斜到1.0。在评估TM-分数和REU之间的关系时,作者观察到一个很强的负相关关系(R2 =−0.67),这表明生成的与天然结构具有相似折叠结构通常会导致较低的Rosetta能量,从而增加了结构的合理性(图3c)。而进一步的实验证明Rosetta最小化确实修复了扩散过程中遇到的一些错误,这无疑有助于ProteinSGM生成高保真的结构。因此,Rosetta最小化可以看作是一个优化步骤,优化结构到更高的质量。
作为一种评估结构可设计性的正交方法,作者使用ProteinMPNN序列设计模型、OmegeFold结构预测网络和scTM评估指标从采样骨干(在快速设计之前使用MinMover)设计序列。实验观察到,scTM > 0.5给出的设计90.2%是可行的,其中可以生成与起始主干具有相同结构的序列(和预测结构)。作者还分析了scTM和预测局部距离差检验(pLDDT)之间的关系。当去除平均pLDDT截止点为70和90的预测结构时-分别对应于自信和非常自信的预测,生成后的样本的scTM比例分别下降到78.3%和24.5%,天然结构分别下降到88.9%和48.3%。作者还评估了scTM和maxTM评分之间的关系,并观察到两者之间存在正相关关系。这表明与训练集相似性较高的结构通常更可设计,这意味着训练集中的蛋白质数据库(PDB)结构都可以通过蛋白质序列实现。
作者还使用DSSP27分析了原生样品和生成样品之间的二级结构分布(图3e,f)。实验发现,与天然结构相比,生成的样本的β-折叠的平均比例略低(分别为0.22和0.24),而α-螺旋的平均比例更高(分别为0.44和0.35)。这符合作者的预期,因为β-折叠需要特定的局部和全局结构约束来形成适当的结构,而α-螺旋更依赖于相邻残基之间的局部相互作用,因此更容易建模。图3h展示了每残基的tm评分为<0.5和REU<-3.5的在训练集中没有发现的新折叠的代表性高保真结构。
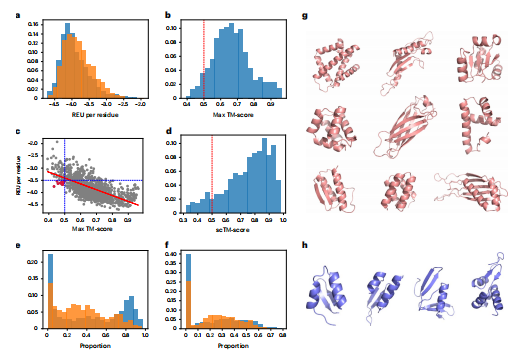
图3 结构分析。用Rosetta最小化、设计和放松了1068个生成的样本,以产生全原子结构。a,与松弛的天然结构相比,该结构的Rosetta能量与所有结构的Rosetta能量有很大的重叠,这表明了结构的可行性.b,将每一次生成的的Maxtm得分与训练集中的所有结构进行比较,作为对训练集中没有发现的新结构的泛化的定量度量。c,Maxtm评分(训练集相似度)和REU(结构可行性)呈负相关,与R2 =−0.67。d,与 ProteinMPNN序列设计模型、OmegaFold和scTM度量的正交分析表明,90.2%的结构(scTM > 0.5)可以通过蛋白质序列实现。e,f,α-螺旋的比例e)和β-折叠(f)在训练集的中生成结构和原始结构之间进行测量。g,h,一些无条件生成的结构的例子(g),包括那些具有新的折叠结构(maxtm-分数< 0.5)和可行的结构(每个残基<-3.5的REU)(h)。
为了确定 ProteinSGM可以产生可折叠的可行骨架,作者选择了一些样本使用圆二色光谱进行实验验证(图4)。总的来说,通过圆二色光谱分析,我们发现蛋白质生成的结构确实可设计、稳定表达、可折叠,并在实验上显示出相似的二级结构组成。
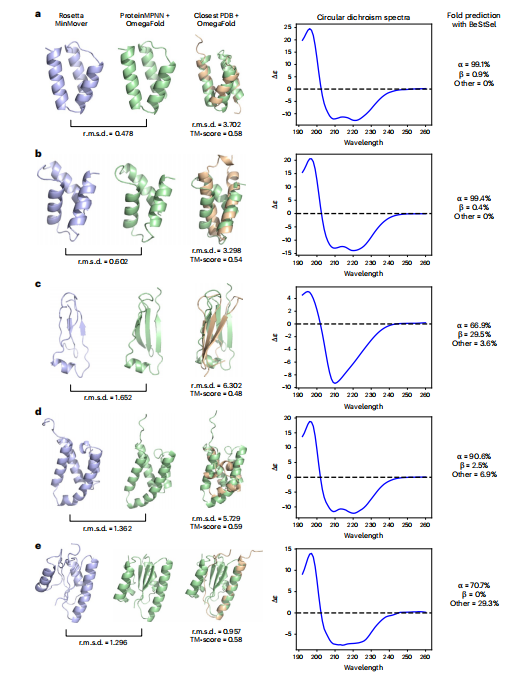
图4 由ProteinSGM,、 ProteinMPNN和OmegaFold生成的无条件样本的实验验证。a-e,作者分析了由α(a,b,d)、αβ (e)和β (c)生成的蛋白,并分析了 Rosetta MinMover(蓝色)、 ProteinMPNN/OmegaFold(绿色)之间的RMSD和tm分数(黄色)。在所有生成的结构中,作者观察到MinMover和OmegeFold结构是一致的(<1.7 A RMSD),与最接近的训练例子有很高的偏差。圆二色光谱和来自圆二色光谱的折叠预测表明,二级结构组成与生成的骨架一致。请注意,尽管e中最接近的训练例子展示了<1 A RMSD,但该结构是一个螺旋环域,与一个螺旋域对齐,但整体拓扑与生成的结构几乎没有相似之处。
4.条件生成
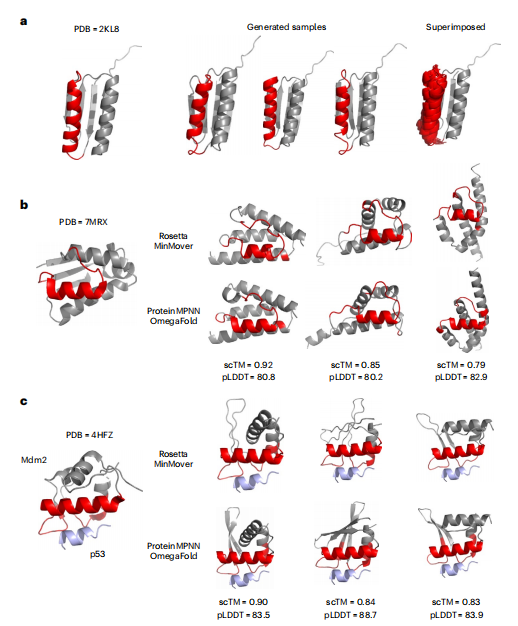
图5 蛋白设计试验案例。a,用红色表示的PDB 2KL8的螺旋结构域被ProteinSGM掩盖和绘制,恢复了原来的螺旋结构。三个随机选择的生成物显示,以及50个随机生成物的叠加,表明y ProteinSGM一致地产生接近天然结构域。b,ProteinSGM可以生成红色的棒星螺旋环结合结构域的从头支架,与天然支架的相似性最小,同时仍然保持螺旋环结构域的一般拓扑结构。c,Mdm2(灰色/红色)通过一个用红色表示的不连续的结构域与p53(蓝色)结合。ProteinSGM可以提出在天然Mdm2支架中不存在的各种αβ折叠的新骨架。
为了解决蛋白质设计中的各种任务,他们训练了一个条件扩散模型,该模型学习如何绘制给定输入结构的任何掩蔽区域。为了评估其性能,作者提出了三个实用的蛋白质设计测试案例的结构域和支架。作者使用了最近发表的一个从头设计的结构(PDB 2KL8),并掩盖了一个长度为20的螺旋域来作为条件扩散模型的输入(图5a)。在所有生成的结构中,该模型在掩蔽区域添加了一个α-螺旋。这表明,该模型已经了解到,鉴于全局结构的约束,一个螺旋可以合理地适合于这个口袋,尽管有轻微的结构差异。这有助于采样具有接近自然拓扑的结构,以优化感兴趣的功能特性,这是蛋白质设计的中心任务。PDB 7MRX代表细菌巴星-巴纳酶复合物,是一种被广泛研究的紧密结合动力学蛋白复合物。Barstar主要通过一个螺旋-环结构域来抑制棒酸酶核糖核酸酶,作者试图为其设计新的骨架(图5b)。模型通过掩蔽蛋白谷氨酸酶Cα距离大于12埃的残基来识别骨架区域。生成的骨架种类多样,Rosetta结构与 ProteinMPNN/OmegaFold结构之间表现出很强的结构一致性,平均pLDDT为>80。这表明生成的骨架在结构上是序列设计的合理性。在另一个测试案例中,作者试图为Mdm2设计骨架,它可以抑制p53肿瘤抑制蛋白(图5c)。同样地,作者屏蔽了p53中Cα距离大于12 A的残基,并使用条件 ProteinSGM模型来绘制掩蔽区域。作者生成了具有高scTM和pLDDT的强候选物,它们保留了与p53的结合位点,但显示了不同的骨架,这表明条件模型适用于各种骨架任务。
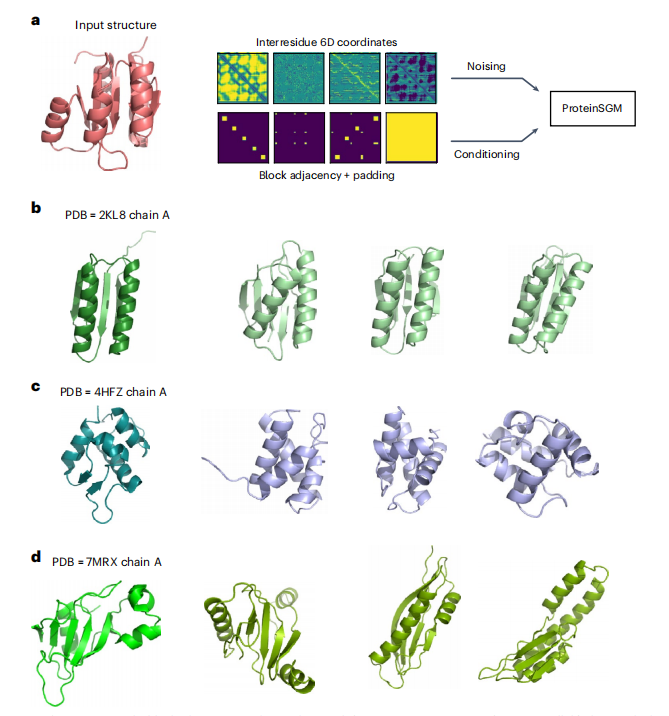
图6 block adjacency conditioning。a,在block adjacency条件模型中,作者从给定的结构中提取了原始的6D坐标特征,并添加了粗略描述蛋白质二级结构特征的块邻接。该模型被训练成以块邻接和长度信息(底部行)为条件的去噪6D坐标(顶部行)。作者提出了三种块邻接条件作用的情况,其中,对于每种情况,作者展示了从中提取块邻接的参考结构,以及由模型生成的三个随机选择的设计。b,以2KL8参考结构为条件的设计在结构上相似但又不同;例如,最左边的结构包含五个反平行的β折叠,这与在参考结构中发现的四个β折叠不同。c,当以螺旋结构为条件时,该模型产生各种螺旋结构,根据螺旋和连接环的排列,这些螺旋结构是唯一可区分的。d,αβ蛋白的条件生成在结构上多样,包括不同的αβ拓扑;然而,请注意,由于二级结构的填充较少,这里的生成物约束较少,因此在块邻接通道中表现出较少的接触约束。
受到block adjacency conditioning的启发,作者提出了一种额外的条件生成方式(图 6)。作者鉴定了由DSSP鉴定的二级结构块,它们对应于>3个连续残基中的α-螺旋或β-片。他们附加了两个额外的通道,对应于α-螺旋和β-sheet块,另一个通道和第三个块接触通道,表明两个块之间的任何Cα坐标是否在7埃内,这会使生成的样本质量变高。
5.结论
ProteinSGM的主要的创新改进包括:(1)提高>256个残基的建模能力;(2)结合多链信息建模蛋白质结合相互作用;(3)用完全可微模块取代Rosetta。目前基于扩散的蛋白质生成的一个主要局限性是,目前的工作仍然依赖于骨架、序列和转子的顺序生成,这限制了蛋白质骨架的自然灵活性,在很大程度上忽略了侧链-主链的相互作用。尽管计算复杂度呈指数级增长,但将这种生成模型扩展到一次性全原子生成是一个很有潜力的有前途的工作途径。
-------------------------------------------
欢迎点赞收藏转发!
下次见!

















 643
643

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









