





《A DNA methylation atlas of normal human cell types》
期刊:Nature
发布时间:2024年4月4日
正常人细胞类型的DNA甲基化图谱
摘要:
DNA甲基化是控制基因表达和染色质组织的基本表观遗传标记,从而为细胞身份和发育过程提供了窗口1.目前的数据集通常只包括一小部分甲基化位点,并且通常基于在培养物中发生巨大变化的细胞系或含有未指定细胞混合物的组织.在这里,我们描述了基于深度全基因组亚硫酸氢盐测序的人类甲基化组图谱,允许对从 205 个健康组织样本中分选的 39 种细胞类型的数千个独特标记物进行片段水平分析。相同细胞类型的重复超过 99.5% 相同,证明了细胞身份程序对环境扰动的稳健性。图谱的无监督聚类概括了组织个体发育的关键要素,并确定了自胚胎发育以来保留的甲基化模式。在单个细胞类型中唯一未甲基化的位点通常位于转录增强子中,并包含组织特异性转录调节因子的 DNA 结合位点。独特的高甲基化位点很少见,并且富集于 CpG 岛、Polycomb 靶标和 CTCF 结合位点,这表明在塑造细胞类型特异性染色质环中具有新作用。该图谱为研究基因调控和疾病相关遗传变异提供了重要资源,并为液体活检提供了大量潜在的组织特异性生物标志物。
介绍
了解相同的DNA序列在不同细胞类型中如何被不同的解释是生物学的一个基本挑战。基因表达、DNA可及性和染色质包装是细胞表型的公认基本决定因素。在这些之下是DNA甲基化,这是一种稳定的表观遗传标记,支撑着细胞身份的终生维持。
现有的人类DNA甲基化数据集存在重大局限性。多项研究表征了胚胎发育、分化、癌症或其他环境的甲基化组依赖于Illumina BeadChip平台,该平台仅限于450,000或860,000个CpG甲基化位点的预定义子集,仅占人类基因组中约3000万个CpG位点的3%.此外,通过独立测量每个 CpG 位点,例如检测忽略了 DNA 甲基化块中发生的 DNA 甲基化的协调模式,这是 DNA 甲基化的关键功能单元.
大多数DNA甲基化分析主要询问大块组织,因此排除了对少数细胞类型的研究,如组织驻留免疫细胞,成纤维细胞或内皮细胞,而其他分析分析则分析培养细胞,其中可能包含体外引入的非生理性甲基化模式.作为部分解决方案,最近的研究使用来自整个组织的单细胞RNA测序数据来鉴定在特定细胞类型中表达的标记基因,然后鉴定其甲基化与表达反相关的特定CpG。这些可用于基于阵列的甲基化组,以对大块组织进行去卷积处理并评估细胞类型组成或样品纯度,但对于鉴定液体活检中的罕见细胞贡献可能不够准确。一些人甲基化组的研究确实使用全基因组亚硫酸氢盐测序(WGBS)分析了分离的原代细胞,但其范围有限2.
为了克服这些局限性并准确表征人类细胞甲基化组,我们对从新鲜解离的成年健康组织中获得的 39 个人类细胞类型组的 39 个人类细胞类型组的荧光激活细胞分选仪 (FACS) 纯化群体进行了深度全基因组测序,平均测序深度为 30×(6.62× 或更高)。我们将整个基因组的甲基化模式合并成**均匀甲基化的CpG位点块,**并用这些位点来研究不同细胞类型甲基化模式的变化。在这里,我们鉴定和表征了以组织或细胞类型特异性方式独特甲基化的基因组区域,提供了其可能的生物学功能的小插曲,并引入了片段级反卷积算法,其应用包括基于循环游离DNA甲基化的临床诊断。
人类细胞类型的甲基化图谱
为了描述各种细胞类型的全基因组DNA甲基化,我们对来自137个同意供体的77种原代细胞类型的205个样本进行了WGBS(150 bp长的双端读长,平均深度至少为30×)。这些被仔细分类并映射到人类基因组(hg19,hg38)。通过流式细胞术、基因表达和 DNA 甲基化分析测定的平均样品纯度(即来自所需细胞类型的材料比例)超过 90%。一些样品的纯度较低(例如,结肠成纤维细胞 78%、平滑肌细胞 (SMC) 82%、内皮细胞 86% 或脂肪细胞 87%)。样品分离和纯度估计的详细说明以及样品信息见补充表1,补充图。**1-3 和补充信息。
分析的细胞类型(图1)。1)代表大多数主要的人类细胞类型**,允许生理系统(例如,胃肠道,造血细胞和胰腺)的综合视图,以及不同环境中相似细胞类型(例如,组织驻留巨噬细胞)的比较。
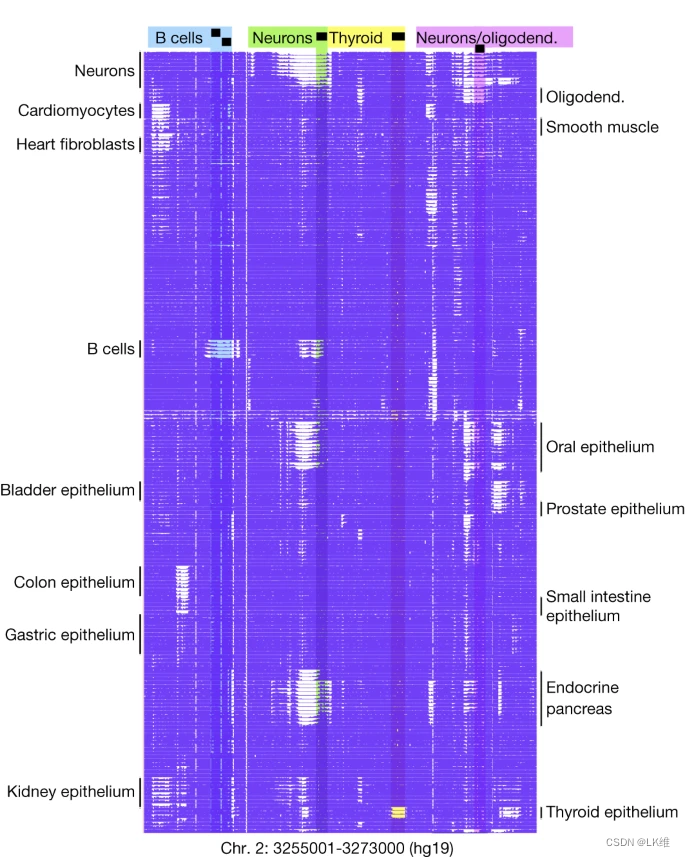
205个甲基化组在重复之间显示出极大的相似性,细胞类型之间以块状方式发生了明显的变化,如图所示。1. 我们试图识别特定细胞类型中差异甲基化的基因组区域,以阐明细胞类型特异性的生物过程,定义细胞身份并促进甲基化生物标志物的开发,以确定循环 cfDNA 片段的细胞来源.
我们开发了 wgbstools,一个计算机器学习套件,用于表示、压缩、可视化和分析 WGBS 数据 (https://github.com/nloyfer/wgbs_tools)。我们通过鉴定多种条件下 DNA 甲基化模式的变化点,将基因组分割成 7,104,162 个不重叠的连续块。每个模块跨越高度相关的 CpG 位点,这些位点在每个样品中甲基化相似,但可能因细胞类型而异(补充信息)。我们保留了至少三个CpG的2,783,421个甲基化块,平均长度为544 bp(四分位距(IQR)= 565 bp)和八个CpG(IQR = 5 CpG)。对这些紧凑的基因组单元进行稳健的分析比单个CpG位点更直接,并且由于甲基化的区域性质,可以将其视为人类DNA甲基化的生物“原子”.
甲基化的个体间差异
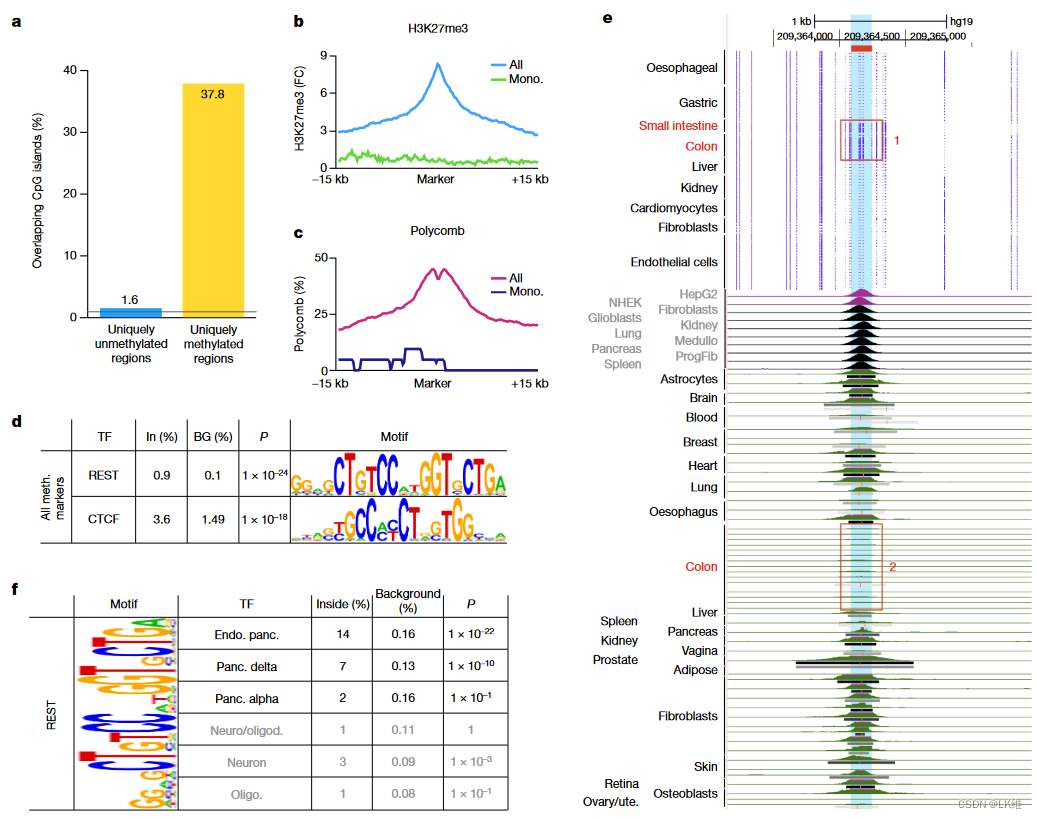
甲基化模式在不同个体中非常稳健。对于大多数细胞类型,0.5%或更少的块在不同供体中显示出50%或更多的差异,而不同细胞类型的样品之间的差异为4.9%(扩展数据图1)。4). 供体之间DNA甲基化的高度相似性与基因组序列的估计个体间变异性相当22.虽然 50% 的定义有些武断,但其他阈值 (35-50%) 显示出类似的趋势,变量块为 0.5% 或更少。在从不同实验室获得的重复中观察到类似的个体间差异(补充表1)。引人注目的是,对于具有 n 个≥ 3 个生物学重复的细胞类型,197 个样本中有 195 个 (99%) 显示出与另一个重复(而不是来自同一供体的另一种细胞类型)的最高相似性。这些结果证明了制剂的可重复性,但也与以前的研究一致6,强调了基本的生物学现象,即DNA甲基化主要由细胞谱系和细胞类型特异性程序决定,而不是由遗传或环境因素决定。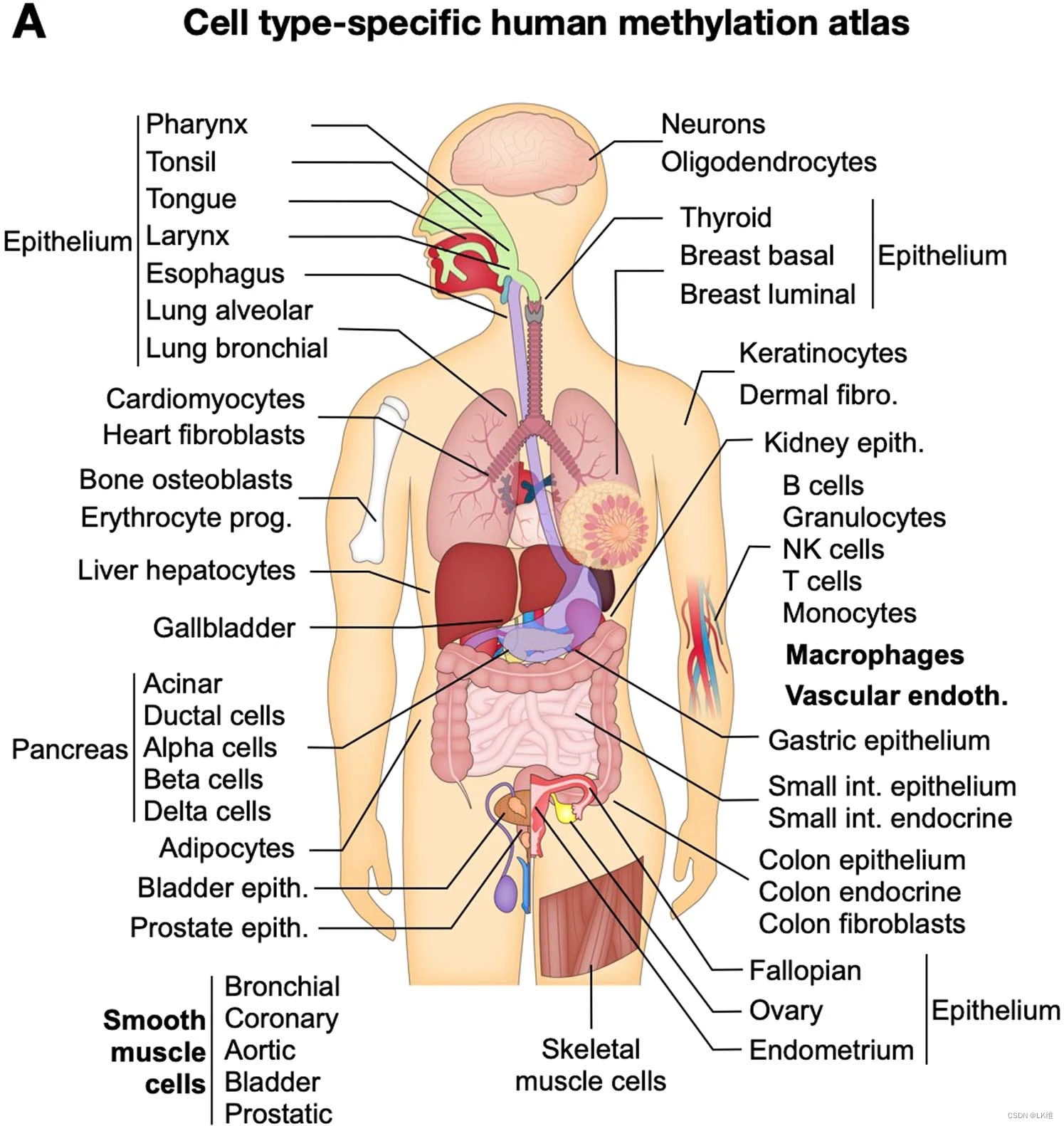
甲基化记录发育史
虽然DNA甲基化模式反映了细胞的功能特性,但它们也可用于跟踪其发育历史。为了确定早期祖细胞后代共享的模式,我们计算了至少四个 CpG 区块内的平均甲基化,并选择了在所有样本中表现出最高变异性的区块(21,000 个区块,前 1%;附表2)。然后,我们使用无监督团聚算法对所有 205 个甲基化组进行聚类,该算法迭代识别并连接两个最接近的样本,而不管它们的标记如何23.该分析系统地对相同细胞类型的生物样品进行分组(图1)。2),类似于纯化的人血细胞的基于阵列的聚类6.这支持了细胞分离的可重复性,并表明每种正常细胞类型的三到四次重复足以推断其甲基化模式,用于生物标志物鉴定等实际应用
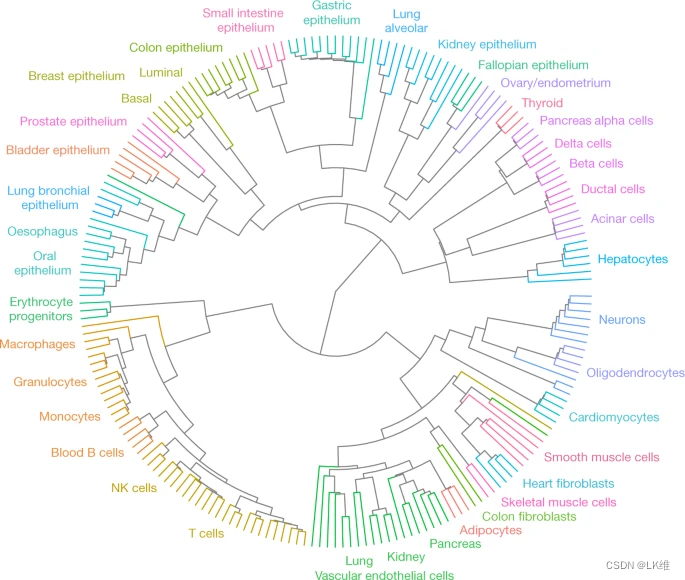
引人注目的是,由此产生的扇形图概括了人体组织之间谱系关系的关键要素。例如,胰岛细胞类型**(α、β 和 delta)**,它们起源于同一胚胎内分泌祖细胞24,密集地聚集在一起。与反映谱系而不是功能的甲基化组一致,胰岛细胞进一步与胰管和腺泡细胞聚集,然后与肝细胞聚集,它们共享内胚层起源。相反,内胚层来源的胰岛细胞不与外胚层来源的神经元聚集25尽管具有常见的组织特异性基因调控和胞吐机制26.
其他例子包括胃、小肠和结肠上皮细胞的聚集;所有血细胞类型的聚类;以及多种中胚层衍生细胞类型的聚集,包括血管内皮细胞、脂肪细胞和骨骼肌。有趣的是,肺支气管上皮与食管和口腔上皮聚集在一起,而肺泡上皮与肠上皮聚集在一起,这与肺泡细胞谱系早期发育起源的证据一致27.
一些甲基化模式在早期发育阶段形成的谱系中很常见。例如,在来源于早期内胚层衍生物的上皮细胞中,有892个区域未甲基化,在中胚层和外胚层来源的细胞中甲基化(方法)。我们认为这些细胞在内胚层胚层中被去甲基化,衍生的细胞类型在几十年后保留了这些模式(扩展数据图1)。由于内胚层衍生物不具有共同的功能或基因表达,这提供了甲基化模式作为稳定谱系标记的另一个例子。
最后,我们将相同的分割和聚类方法应用于路线图表观基因组学项目中已发表的甲基化图谱4.该算法没有对相关的细胞类型进行分组,并且通常根据供体身份对样本进行聚类。这进一步强调了仔细纯化均质细胞类型的重要性,避免混合细胞群(扩展数据图1)。5b)。
细胞类型特异性甲基化标志物
接下来,我们转向研究以细胞类型特异性方式差异甲基化的基因组区域。我们将 205 个样本分为 39 组特定细胞类型,包括血细胞类型**(B、T、自然杀伤 (NK)、粒细胞、单核细胞和组织驻留巨噬细胞)**、乳腺上皮(基底和管腔)、肺上皮(肺泡和支气管)、胰腺内分泌(α、β 和 δ)和外分泌(腺泡和导管)细胞、各种来源的血管内皮细胞、心肌细胞和心脏成纤维细胞等。我们还定义了 12 个超组,其中对相关细胞类型进行了分组,包括肌肉细胞、胃肠上皮细胞、胰腺等(补充表 3)。
然后,我们专注于由五种或更多CpG组成的差异甲基化块,这些CpG在一组细胞类型中未甲基化,但在所有其他样品中甲基化,反之亦然。有趣的是,几乎所有区域(97%)在一种细胞类型中都是非甲基化的,而在所有其他细胞类型中都是甲基化的。然后,我们根据靶细胞类型与所有其他样品的甲基化绝对差异对这些差异区域进行排序(方法和补充信息)。
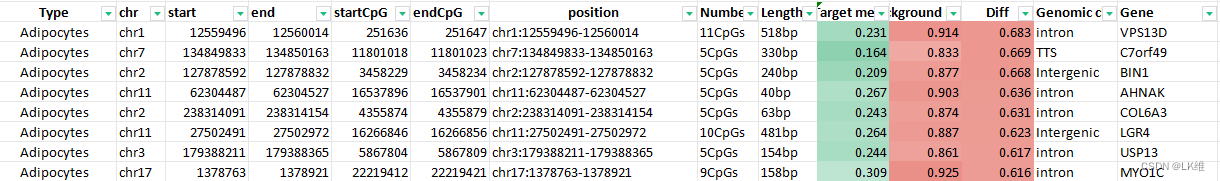
每种细胞类型的前 25 个差异非甲基化区域包括 1,246 个标记物的人类细胞类型特异性甲基化图谱(图 1)。3和附表4)。
附表4
| Type | chr | start | end | startCpG | endCpG | position | Number of CpGs | Length | Target meth. | Background meth. | Diff | Genomic class | Gene |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Adipocytes | chr7 | 134849833 | 134850163 | 11801018 | 11801023 | chr7:134849833-134850163 | 5CpGs | 330bp | 0.164 | 0.833 | 0.669 | TTS | C7orf49 |
| Adipocytes | chr11 | 62304487 | 62304527 | 16537896 | 16537901 | chr11:62304487-62304527 | 5CpGs | 40bp | 0.267 | 0.903 | 0.636 | intron | AHNAK |
| Adipocytes | chr11 | 27502491 | 27502972 | 16266846 | 16266856 | chr11:27502491-27502972 | 10CpGs | 481bp | 0.264 | 0.887 | 0.623 | Intergenic | LGR4 |
| … | … | … | … | … | … | … | … | … | … | … | … | … | … |
人类细胞类型特异性调控图谱
接下来,我们转向表征这些细胞类型特异性的差异非甲基化区域。为此,我们确定了每种细胞类型的前****250个未甲基化标记(补充表4b),
并使用GREAT来识别与每组标记相邻的基因
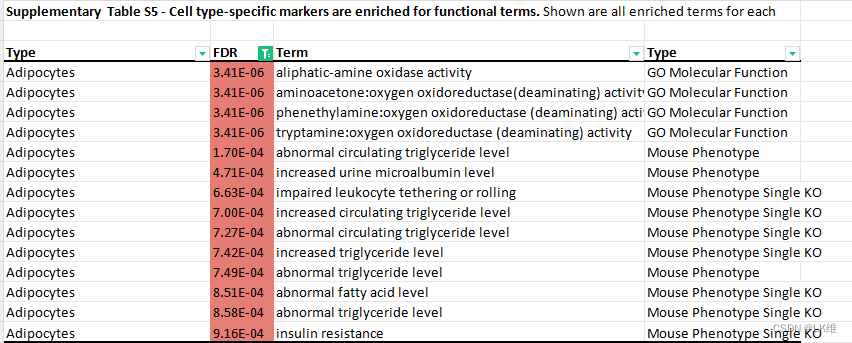
,并测试它们对各种基因集注释的富集31.在给定细胞类型中唯一未甲基化的基因座相邻的基因通常反映了该细胞类型的功能特性。例如,B细胞标志物附近的基因富集了B细胞形态、分化、IgM水平和淋巴细胞生成;NK细胞标志物与NK细胞介导的细胞毒性、造血系统、细胞毒性和淋巴细胞生理学相关;输卵管标志物富集于蛋壳和卵黄周围空间;以及心脏松弛、收缩压、肌肉发育和肥大的心肌细胞标志物(补充表5)。
然后,我们分析了细胞类型特异性标记物的 DNA 可及性和染色质包装,如使用测序 (ATAC-seq)、DNase I 超敏位点测序 (DNaseI–seq) 测定转座酶可及染色质所定义4,32和组蛋白标记指示活性启动子和增强子4.单核细胞和巨噬细胞的前 250 个未甲基化标志物高度可及,并以单核细胞中的 H3K27ac 和 H3K4me1 为特征,而其他细胞类型的标志物在单核细胞中没有富集(图 1)。4a),其他细胞类型的标志物也有类似的结果(扩展数据图4a)。7). 我们还显示出 chromHMM 增强子注释在细胞类型特异性标记物上的强烈协调富集33(图。这些发现与先前的研究一致,这些研究将组织特异性去甲基化与基因增强子相关联1,34.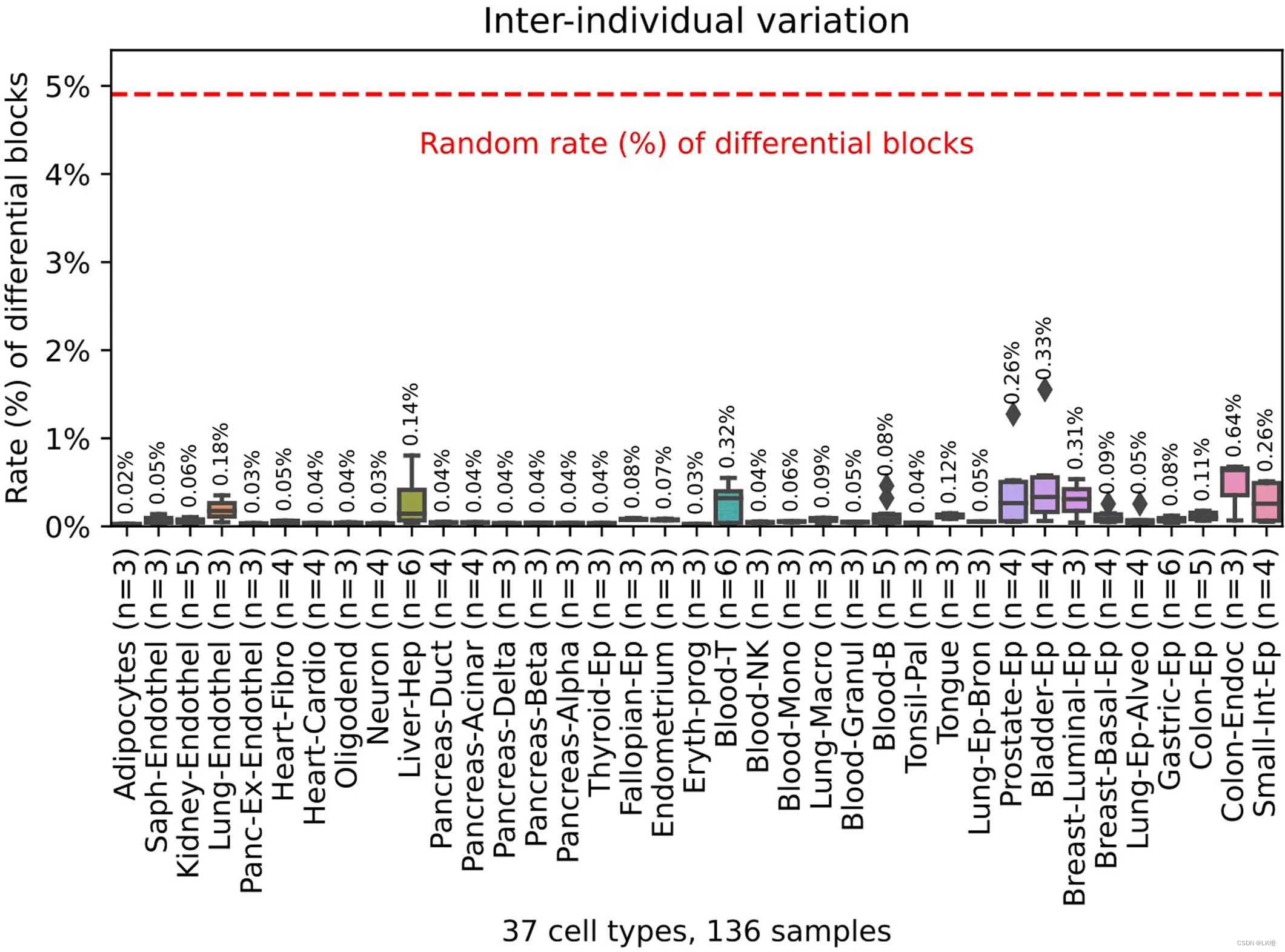
为了进一步评估细胞类型特异性未甲基化区域的生物学重要性,我们研究了它们与转录因子(TFs)的关联,这些转录因子可能影响DNA甲基化或根据甲基化和染色质以细胞类型特异性的方式结合DNA。我们鉴定了每种细胞类型的前1000个未甲基化标记(补充表4c),并使用HOMER进行基序分析,以计算已知转录因子结合基序的富集(补充表6a)。对于大多数细胞类型,顶级基序包括主调控因子和关键转录因子(图4b)。例如,B细胞富集于Ebf2/HEB/E2A,粒细胞富集于CEBP/AP1/ETS,T细胞富集于ETS/RUNX。细胞类型特异性未甲基化区域与转录因子结合基序之间的关联可以鉴定新的基因调控回路,并暴露在特定细胞类型中活跃的远端增强子。
我们旨在鉴定由细胞类型特异性去甲基化标记的可能增强子的目标基因。顶级标记经常落在内含子区域之内,并可能调节这些基因(例如,胰岛α细胞中的胰高血糖素,心肌细胞中的NPPA和MYL4,以及少突胶质细胞中的MBP;补充表7),或接近可能的目标(例如,胰岛素基因5kb处的β细胞标记)。其他标记与其目标基因距离较远。我们设计了一个计算算法,以鉴定在匹配条件下细胞类型特异性标记附近的基因表达水平增加的基因(方法)。这突出了许多细胞类型的标志基因,并为每种细胞类型的前25个未甲基化标记提出了许多假定目标。例如,肝细胞标记与APOE、APOC1、APOC2和胰高血糖素受体相关。同样,心肌细胞标记与NPPA、NPPB和肌球蛋白基因相关;胰岛标记与胰岛素和胰高血糖素基因相关(补充表7)。这些发现进一步支持了这样一个原则:在特定细胞类型中特异性未甲基化的位点可能是正向调节该细胞类型中表达基因的增强子,通常控制相邻基因。然而,我们注意到,在特定细胞类型中与特异性未甲基化位点相邻的基因通常在这一细胞类型之外广泛表达(讨论)。为了在每种细胞类型中生成一个假定调控区域的目录,我们在不考虑其他细胞类型的情况下,对每种细胞类型的所有样本进行了片段级分析。我们扫描了整个基因组,并鉴定了至少85%的DNA片段(至少有四个CpG)未甲基化的基因组区域(方法)。这在分析的39个细胞类型组中鉴定了一组未甲基化的基因组区域,平均每组有36111个区域(补充数据集1)。然后对这些区域进行了基因组特征注释,显示平均有56%与CpG岛重叠,46%靠近启动子区域,44%与CTCF结合位点重叠,从而突出了未甲基化位点的调控和结构角色。当可用时,我们将这些区域与ENCODE和Roadmap Epigenomics在匹配条件下的染色质免疫沉淀测序(ChIP-seq)峰进行了交叉对比,包括H3K4me3、H3K27ac、H3K4me1、H3K27me3、CTCF和ATAC-seq,并生成了一个由未甲基化区域组成的细胞类型特异性假定增强子区域目录,这些区域与H3K27ac但不与H3K4me3峰重叠(补充数据集2)。这些区域的基序分析鉴定了每种细胞类型中的关键转录因子,类似于图4中显示的那些(补充表6b,c)。
细胞类型特异性高甲基化位点
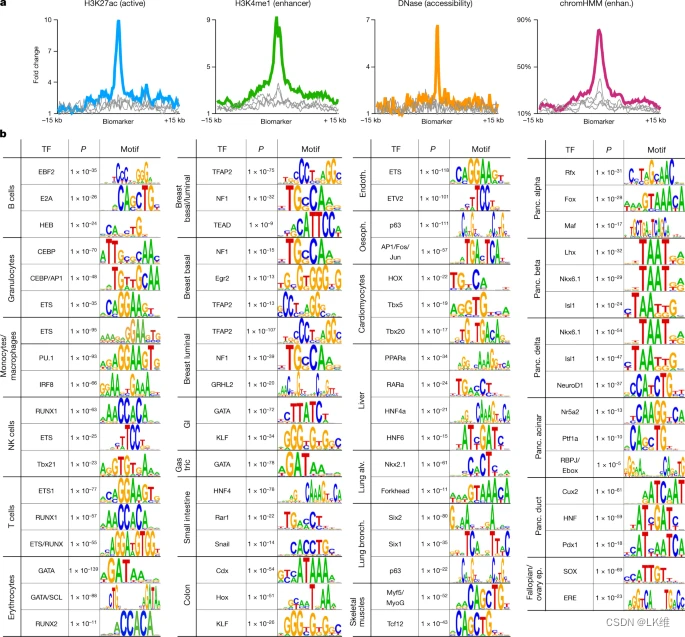
我们研究了那些在一种细胞类型中甲基化但在人体其他部位未甲基化的基因组区域。这些富集用于 CpG 岛(38% 的甲基化区域,而细胞类型特异性未甲基化区域为 1.7-2.7%),并且在其他细胞类型中被 H3K27me3 和 Polycomb 标记(图 1)。5a-c),如先前报道的癌症和发育过程40,41.这些细胞类型特异性的高甲基化区域通常对基序富集不那么显著(与独特的未甲基化区域相比)。有趣的是,只有大约3%的细胞类型特异性差异甲基化区域是高甲基化的。
在汇集所有细胞类型特异性高甲基化区域后,我们鉴定了染色质调节因子CTCF靶序列的强富集(P ≤ 1 × 10–18;无花果。这表明CTCF结合位点的DNA甲基化可以作为组织特异性调节开关来调节其结合,从而可能影响组织特异性三维基因组组织35,42,43.为了验证这一想法,我们将 CTCF 位点的 DNA 甲基化模式与特定组织中的全基因组 CTCF 蛋白结合进行了比较。图5e显示了甲基化模式和在一个基因座上公布的体内CTCF占有率,该基因座在结肠和肠道中特异性甲基化。与阻止 **CTCF 结合的 DNA 甲基化一致,**ChIP 数据显示结肠中该位点选择性地不存在 CTCF 结合。此外,在特定细胞类型中甲基化的位点富集了神经基因转录抑制因子RE1沉默TF/神经元限制性沉默因子(REST/NRSF)的靶标(P ≤ 1 × 10–24),这在胰岛细胞的甲基化组中最为明显(图1)。虽然DNA甲基化尚未被证明影响REST的结合或活性,但这一发现提出了一种有趣的可能性,即胰岛中REST靶标的甲基化可以独立于REST抑制而允许内分泌分化。
片段级甲基化组反卷积
最后,我们开发了一种用于 DNA 甲基化测序数据的计算片段级反卷积算法,并使用为每种细胞类型定义的前 25 个标记(总共 1,246 个标记)来研究从复合组织样品和 cfDNA 中获得的甲基化组。简而言之,我们生成了一个图谱,其中计算了每种细胞类型(列)中每个标记(行)的未甲基化片段的百分比。然后使用非负最小二乘法 (NNLS) 算法来拟合输入样本并估计其相对贡献(补充信息)。
为了估计片段级方法的准确性,我们使用了测序读长的计算机混合物。对于每种细胞类型,我们应用留一方法在白细胞读数中混合一个保留的样品,然后使用反卷积算法来推断混合物中的细胞组成。我们在0到10%的浓度下重复这个过程。如图所示。6a,我们发现 1,246 个标记(每种细胞类型的前 25 个)允许以大约 0.1% 的分辨率准确检测来自给定来源的 DNA,与基于阵列的方法相比,提高了近一个数量级28.四元计算机混合,其中还包括内皮细胞和肝细胞甲基化组以真实地模拟cfDNA组成,产生了类似的结果(扩展数据图1)。8).
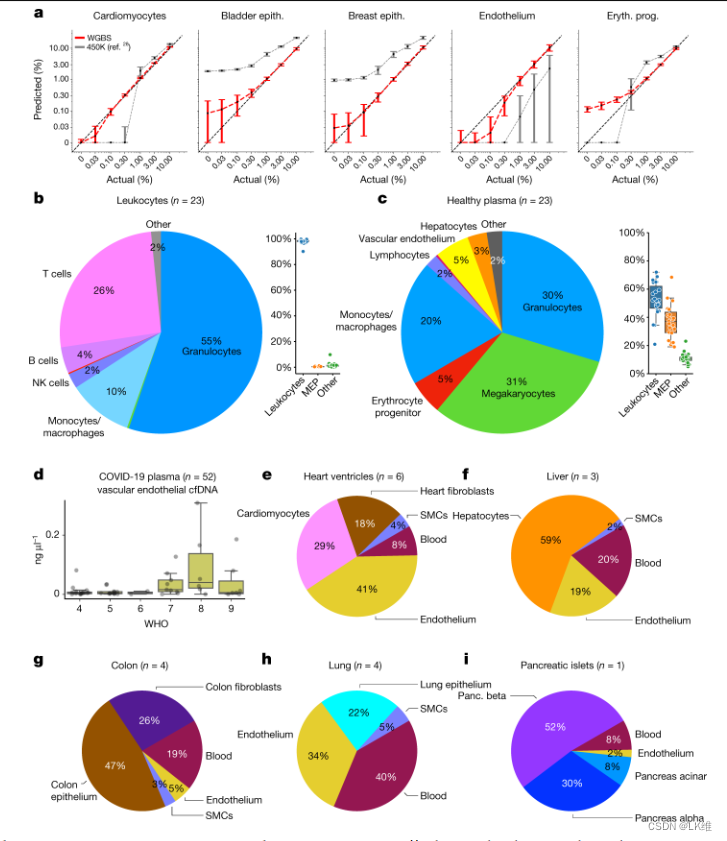
然后,我们使用来自 23 名健康供体的 WGBS 数据估计了白细胞和 cfDNA 的细胞组成;99.5%的白细胞来源的DNA归因于粒细胞、单核细胞、巨噬细胞和NK、T和B细胞,与典型血细胞计数一致(图1)。6b和补充表8)。健康受试者的cfDNA主要来源于白细胞:粒细胞(29.7%)、单核细胞/巨噬细胞(20%)和淋巴细胞(3%)。贡献cfDNA的实体组织包括血管内皮细胞(6%)和肝细胞(3.1%)(图1)。6c),与之前的结果一致28.目前的图谱还显示巨核细胞(31%)和红细胞祖细胞(prog.)细胞(5%)对cfDNA有显着贡献,这在以前使用范围更有限的参考甲基化组的研究中没有观察到。
COVID-19 患者的内皮 cfDNA
基于DNA甲基化模式的分析为鉴定cfDNA的组织来源提供了机会。COVID-19 会对多个组织造成损害,其中一些组织没有生物标志物。我们使用该图谱对 52 名因 COVID-19 住院的患者的浅层 WGBS 数据进行反卷积(参考文献)。44).我们从粒细胞、红细胞祖细胞、肺和肝脏中鉴定出过多的游离DNA片段,这与已发表的这些样本的分析一致(补充信息)。引人注目的是,我们还发现了血管内皮细胞对这些患者的cfDNA的显着贡献,在没有内皮细胞甲基化组参考的情况下,在已发表的分析中无法检测到这一点(图1)。有趣的是,与病情较轻的患者(WHO评分≥≤6;第 6 页≤ × 10–5,曼恩-惠特尼)。这些结果表明,血管内皮细胞死亡在 COVID-19 的发病机制中起着重要作用,可能与凝血功能障碍有关,并强调了使用全面的细胞类型特异性图谱进行 cfDNA 甲基化组分析的好处。
复合组织的细胞类型反卷积
最后,我们分析了ENCODE的全基因组甲基化组5和路线图表观基因组学图谱4使用我们的图谱(基于每种细胞类型的 25 个标记)。一些甲基化组的反卷积显示出预期的均质组成,例如,路线图T细胞样品中97-99%的T细胞DNA(补充表9)。然而,对其他样品的分析显示出高度异质的组成,正如之前基于基于阵列的散装组织反卷积算法(如 EpiDISH 和 EpiScore)所报道的那样14,15,45.例如,心室样本包括 29% 的心肌细胞、41% 的内皮细胞和 18% 的心脏成纤维细胞(图 1)。6e);肝脏甲基化组包括约60%的肝细胞、21%的血液和20%的内皮细胞;结肠甲基化组包括约50%的结肠上皮细胞、26%的结肠成纤维细胞和19%的血液。最引人注目的是,路线图肺样本以血液(40%)、内皮(34%)和平滑肌(5%)为主,只有22%的DNA来自肺上皮细胞(图1)。6f-i和补充表9)。重要的是,对此处介绍的 205 个样品进行类似的反卷积,对每个样品的预期细胞类型的平均贡献为 94%(中位数为 95%,补充表 10),或在更严格的留一交叉验证分析中为 91%(中位数为 92%)(补充表 11),突出了收集样品的纯度。
当然,片段级分析仅限于具有全基因组测序数据的细胞类型,并且某些细胞类型只能通过基于阵列的算法进行分析15,28.尽管如此,这里介绍的标记物和算法允许对复合体组织和血浆样品进行分析,跨越多种细胞类型,并且具有很高的准确性。
讨论
本文描述的人类细胞类型甲基化组的综合图谱阐明了 DNA 甲基化的原理,并为多条研究线以及转化应用提供了宝贵的资源。
我们的分析使用全基因组测序数据表明,甲基化模式在来自不同个体的相同细胞类型的健康重复中惊人地相似。个体之间的相似性反映了细胞分化和维持回路的稳健性,至少就健康组织而言是这样。涉及表观基因组不稳定的病理学显然会破坏这些回路,导致来自特定正常细胞类型的细胞之间出现更多种类的甲基化模式。我们预测,即使在癌症(具有相同的原发解剖部位和组织学类型)中,在甲基化阻滞水平上对纯化的上皮细胞进行比较甲基化组分析,也将显示出比通常假设的更小的个体间差异。
正如图谱所示,每种细胞类型都有一组基因组区域,与其他细胞类型相比,这些基因组区域在该细胞类型中是唯一的未甲基化区域,以及与相关细胞类型共享甲基化模式的其他基因组区域。使用细胞类型特异性甲基化组的无监督聚类,我们发现细胞类型的聚类方式反映了其发育起源而不是表达模式。这提供了一个引人入胜的观点,即DNA甲基化是祖细胞甲基化的记录,通过戏剧性的发育转变和此后几十年的生命保留在基因组中。我们提出,比较甲基化组分析将允许重建胎儿结构或细胞类型的部分甲基化组,类似于进化生物学中最后一个共同祖先的重建。
绝大多数细胞类型特异性差异甲基化区域在一种细胞类型中被特异性去甲基化。这些区域的染色质通常很容易获得,并带有与活性基因调控相关的组蛋白标记,如在增强子和启动子中发现的那样。此外,这些位点富集了在该细胞类型中起作用的 TF 结合位点基序。我们设计了一种综合方法,基于距离和基因表达谱,使我们能够突出这些假定增强子区域的潜在靶基因。许多增强子区域与附近广泛表达的基因相关,可能反映了多种组织特异性增强子的基因调控。我们的研究结果与先前的研究结果一致,这些研究显示组织特异性低甲基化发生在基因增强子处.我们的数据驱动标记鉴定方法是对最近以基因为中心的方法的补充14,15使用组织特异性单细胞 RNA 测序数据来定义标记基因并鉴定在靶细胞类型中特异性未甲基化的相邻 CpG。最后,我们设计了一种片段水平的基因组分析,以鉴定每种细胞类型的数以万计的未甲基化区域,这些区域用基因组特征、DNA 可及性、染色质标记和 TF 结合基序进行注释,以产生推定增强子的细胞类型特异性目录。对该图谱的进一步分析将显示并验证每种细胞类型中的完整人类增强子集。
相反,我们鉴定了一种或两种细胞类型中特异性甲基化的基因组区域,约占细胞类型特异性差异甲基化区域的 3%。它们通常位于 CpG 岛中,其特征在于 H3K27me3 和 Polycomb 结合在基因座未甲基化的组织中.这种表观遗传抑制转换以前在癌症和早期发育过程中被描述过41,46,但其在特定细胞类型分化中的作用仍不清楚。这些区域富集了 CTCF 结合位点,表明 DNA 甲基化在减弱 CTCF 结合中的作用,从而调节相邻 DNA 的细胞类型特异性三维组织35,36,47.
对于DNA甲基化测序数据,据我们所知,这里描述的图谱是迄今为止最全面的纲要。我们鉴定了一千多个细胞类型独特的DNA甲基化区域,这些区域可以作为准确和特异性的生物标志物,用于片段水平分析和通过监测cfDNA来识别细胞死亡事件。值得注意的是,这些标记区域中的大多数没有被 450K/EPIC BeadChip DNA 甲基化阵列覆盖,并且以前没有得到重视。为了能够解释阵列数据,我们提供了一组替代的细胞类型特异性标记物,仅限于 BeadChip 450K 阵列中包含的 CpG 位点。同样,我们在RRBS和杂交捕获panel靶向区域中鉴定了细胞类型特异性标记物(扩展数据图1)。9和附表12-17)。如扩展数据图所示。10、阵列适应图谱允许对胰岛、肺和乳腺活检的阵列甲基化组进行高分辨率解释,突出显示以前未分析的细胞类型的存在48,49,50.
图谱中缺少许多细胞类型,通常是因为材料的可用性有限。例子包括成骨细胞、胆管细胞、肾上腺细胞、尿道上皮细胞和造血干细胞。此外,我们没有分离出许多感兴趣的亚群,例如不同类型的神经元或淋巴细胞。该地图集被视为一个活生生的、公开的数据库,将来会更新。图谱的分辨率产生了对复合组织的定量理解,并允许人们识别尚未表征的其他细胞类型的缺失甲基化组。我们还承认,由于用于FACS的抗体的质量以及它们允许分离细胞类型的程度不同,分选细胞群的纯度各不相同。尽管如此,即使是图谱中纯度最低的细胞类型(例如,一些血管内皮细胞、成纤维细胞、SMC 和脂肪细胞的制剂,纯度为 70-80%),当平均重复时,也可用于鉴定差异甲基化区域和推断混合物中的细胞组成。
总之,我们提供了一份全面的原代人类细胞类型的甲基化图谱,以及一套广泛的细胞类型特异性标记物和计算工具,用于混合细胞类型样品的片段级分析。这些补充了大量可用于分析阵列数据的基于阵列的甲基化组和反卷积工具。总之,这些数据揭示了DNA甲基化在细胞生物学和基因调控中的作用,并有助于鉴定在每种细胞类型中活跃的增强子。也许我们的图谱最有前途的用途是混合细胞类型样本的片段水平反卷积的潜力,从而可以灵敏地鉴定癌症和其他疾病患者血浆中cfDNA的起源组织.
问题:
- 标准化如何做
- 附表4中Target meth. 和Background meth.如何获取

















 516
516

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









