







文章复现:多组学微生物大分子相互作用 Fig.4d-Sankey Plot
赠送代码方式:关注桓峰基因发消息'ASD'获得下载链接!!
文章基本信息
标题: Integrative multi-omics analysis of autism spectrum disorder reveals unique microbial macromolecules interactions
作者: Osama A,et al.
期刊: J Adv Res
发表时间: 2025 Jan
影响因子: 11.4
DOI: doi: 10.1016/j.jare.2025.01.036
文章复现: Fig.4d-Sankey Plot
摘要
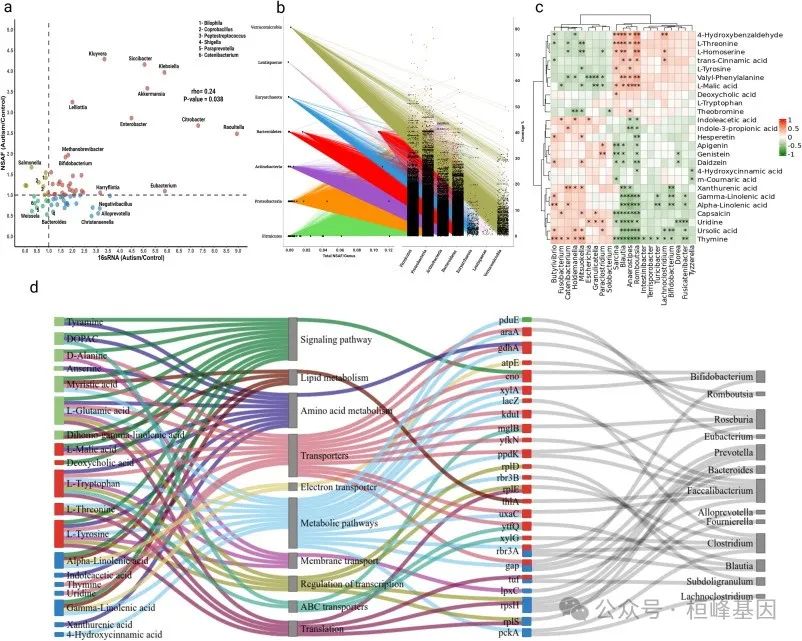
介绍:肠道微生物群的改变与自闭症谱系障碍(ASD)有关,但将这些变化与ASD病理生理联系起来的机制尚不清楚。
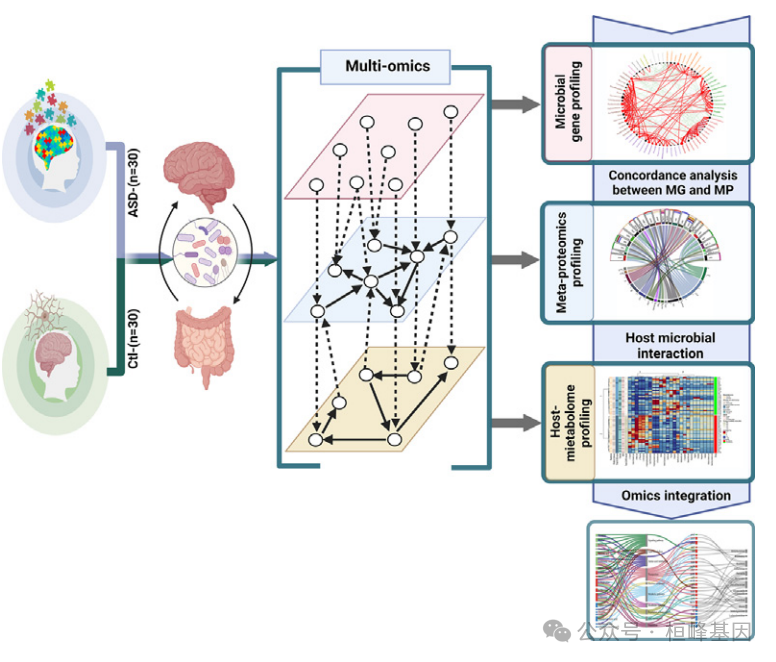
目标:本研究利用多组学方法,通过检测微生物多样性、细菌元蛋白、相关代谢途径和宿主蛋白质组,揭示肠道微生物群与ASD之间的联系机制。
方法:分析了30例重度ASD患儿和30例健康对照者的肠道微生物群。采用16S rRNA V3和V4测序评估微生物多样性。一种新的宏蛋白质组学管道识别细菌蛋白质,而非靶向代谢组学探索改变的代谢途径。最后,采用多组学整合将大分子变化与神经发育缺陷联系起来。
结果:与对照组相比,患有ASD的儿童肠道微生物群发生了显著变化,包括多样性和丰富度较低。tyzzerella与ASD组有独特的联系。微生物网络分析显示ASD的重新布线和稳定性降低。鉴定的主要超蛋白由双歧杆菌和克雷伯菌产生(如木糖异构酶和NADH过氧化物酶)。代谢组学分析鉴定出神经递质(如谷氨酸、多巴胺)、脂质和氨基酸能够穿过血脑屏障,可能有助于神经发育和免疫失调。宿主蛋白质组学分析显示,参与神经炎症和免疫调节的蛋白发生了改变,包括钾化因子(KLK1)和转甲状腺素(TTR)。最后,多组学整合支持单组学研究结果,并加强了肠道微生物群及其大分子产物可能与ASD相关症状有关的假设。
结论:多组学数据的整合提供了关键证据,表明肠道微生物群的改变和相关大分子的产生可能在ASD相关症状和合并症中发挥作用。关键细菌超蛋白和代谢物被确定为ASD神经和免疫失调的潜在贡献者,强调了治疗干预的可能新靶点。
软件包安装
if(!require(networkD3))
install.packages ("htmlwidgets")
if(!require(networkD3))
install.packages ("networkD3")数据读取
准备links
library(networkD3)
library(dplyr)##
## 载入程序包:'dplyr'## The following objects are masked from 'package:stats':
##
## filter, lag## The following objects are masked from 'package:base':
##
## intersect, setdiff, setequal, unionlinks <- readxl::read_excel("Sankey plot.xlsx", sheet = 4)
colnames(links) <- c("target", "source", "group")
links$value <- 1
links$group <- as.factor(links$group)
head(links)## # A tibble: 6 × 4
## target source group value
## <chr> <chr> <fct> <dbl>
## 1 araA Metabolic pathways type_Metabolic pathways 1
## 2 araA Transporters type_Transporters 1
## 3 atpE Electron transporter type_Electron transporter 1
## 4 eno Metabolic pathways type_Metabolic pathways 1
## 5 eno Signaling pathway type_Signaling pathway 1
## 6 eno Transporters type_Transporters 1准备nodes
# From these flows we need to create a node data frame: it lists every entities involved in the flow
nodes <- data.frame(
name=c(as.character(links$source), as.character(links$target)) %>%
unique()
)
# With networkD3, connection must be provided using id, not using real name like in the links dataframe.. So we need to reformat it.
links$IDsource <- match(links$source, nodes$name)-1
links$IDtarget <- match(links$target, nodes$name)-1
nodes$group <- as.factor(c("my_unique_group"))
head(nodes)## name group
## 1 Metabolic pathways my_unique_group
## 2 Transporters my_unique_group
## 3 Electron transporter my_unique_group
## 4 Signaling pathway my_unique_group
## 5 Amino acid metabolism my_unique_group
## 6 Membrane transport my_unique_group配置颜色
# prepare color scale: I give one specific color for each node.
pathways <- links
pathways <- pathways$group
pathways <- unique(pathways)
pathways <- as.character(pathways)
pathways <- pathways[!pathways %in% ""]
pathways <- paste0("type_" , pathways)
pathways## [1] "type_type_Metabolic pathways"
## [2] "type_type_Transporters"
## [3] "type_type_Electron transporter"
## [4] "type_type_Signaling pathway"
## [5] "type_type_Amino acid metabolism"
## [6] "type_type_Membrane transport"
## [7] "type_type_ABC transporters"
## [8] "type_type_Regulation of transcription"
## [9] "type_type_Translation"
## [10] "type_type_Lipid metabolism"
## [11] "type_NA"my_color_color <- paste0("d3.scaleOrdinal() .domain([")
pathways <- paste0("'", pathways , "'")
pathways <- paste0(pathways, collapse = ",")
my_color_color <- paste0(my_color_color , pathways , ",'my_unique_group'" , "]) .range([" )
head(my_color_color)## [1] "d3.scaleOrdinal() .domain(['type_type_Metabolic pathways','type_type_Transporters','type_type_Electron transporter','type_type_Signaling pathway','type_type_Amino acid metabolism','type_type_Membrane transport','type_type_ABC transporters','type_type_Regulation of transcription','type_type_Translation','type_type_Lipid metabolism','type_NA','my_unique_group']) .range(["## install.packages("remotes")
#remotes::install_github("Nowosad/rcartocolor")
library(rcartocolor)
colors <- c(carto_pal(11, "Safe") , "grey")
colors <- paste0("'", colors , "'")
colors <- paste0(colors, collapse = ",")
my_color_color <- paste0(my_color_color, colors , "])" )
my_color_color## [1] "d3.scaleOrdinal() .domain(['type_type_Metabolic pathways','type_type_Transporters','type_type_Electron transporter','type_type_Signaling pathway','type_type_Amino acid metabolism','type_type_Membrane transport','type_type_ABC transporters','type_type_Regulation of transcription','type_type_Translation','type_type_Lipid metabolism','type_NA','my_unique_group']) .range(['#88CCEE','#CC6677','#DDCC77','#117733','#332288','#AA4499','#44AA99','#999933','#882255','#661100','#888888','grey'])"绘图sankey图
# Make the Network. I call my colour scale with the colourScale argument
p <- sankeyNetwork(Links = links,
Nodes = nodes,
Source = "IDsource",
Target = "IDtarget",
Value = "value",
NodeID = "name",
colourScale = my_color_color,
sinksRight = T ,
fontSize = 17 ,
LinkGroup="group",
NodeGroup="group")## Links is a tbl_df. Converting to a plain data frame.图形保存
library(htmlwidgets)##
## 载入程序包:'htmlwidgets'## The following object is masked from 'package:networkD3':
##
## JSsaveNetwork(p, "sankey Final.html")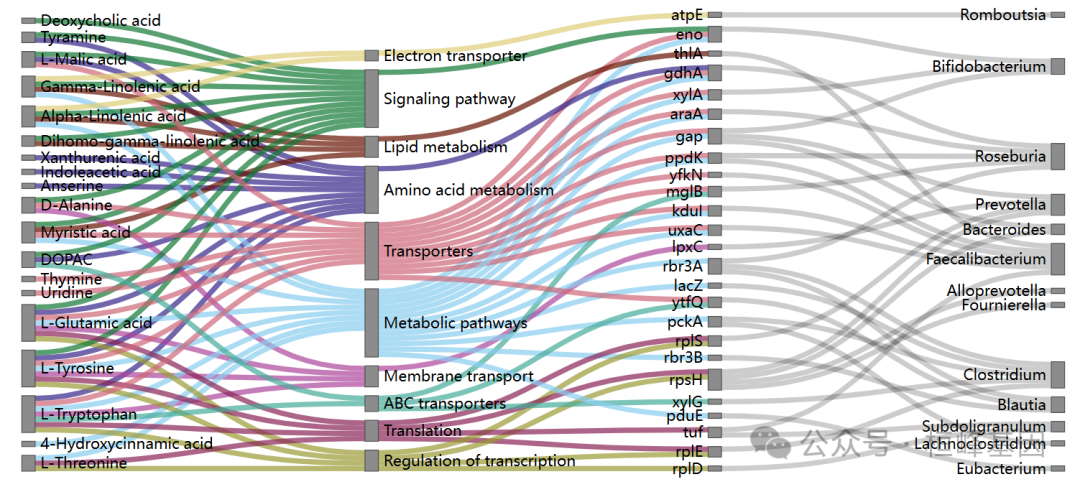
Reference
Osama A, et al. Integrative multi-omics analysis of autism spectrum disorder reveals unique microbial macromolecules interactions. J Adv Res. 2025 Jan 25:S2090-1232(25)00055-4.


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









